Article Text

Abstract
Cluster randomised clinical trials present unique challenges in meeting ethical obligations to those who are treated at a randomised site. Obtaining informed consent for research within the context of clinical care is one such challenge. In order to solve this problem it is important that an informed consent process be effective and efficient, and that it does not impede the research or the healthcare. The innovative approach to informed consent employed in the COMPASS study demonstrates the feasibility of upholding ethical standards without imposing undue burden on clinical workflows, staff members or patients who may participate in the research by virtue of their presence in a cluster randomised facility. The COMPASS study included 40 randomised sites and compared the effectiveness of a postacute stroke intervention with standard care. Each site provided either the comprehensive postacute stroke intervention or standard care according to the randomisation assignment. Working together, the study team, institutional review board and members of the community designed an ethically appropriate and operationally reasonable consent process which was carried out successfully at all randomised sites. This achievement is noteworthy because it demonstrates how to effectively conduct appropriate informed consent in cluster randomised trials, and because it provides a model that can easily be adapted for other pragmatic studies. With this innovative approach to informed consent, patients have access to the information they need about research occurring where they are seeking care, and medical researchers can conduct their studies without ethical concerns or unreasonable logistical impediments.
Trial registration number NCT02588664, recruiting. This article covers the development of consent process that is currentlty being employed in the study.
- informed consent
- research ethics
- ethics committees/consultation
- clinical trials
- regulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The central intent of the informed consent process is to protect the rights of individuals to autonomy by enabling them to decide whether or not to participate in research, based on information they receive about the potential risks and benefits offered by the study, and the activities they will participate in if they voluntarily enrol.1–3 Significant questions remain about ethical conduct of pragmatic clinical trials (PCT) in fast-paced clinical settings.4–8 There may be an inherent assumption that research activities of learning healthcare systems (LHS) seamlessly integrate with clinical care and comply with the rigorous, predetermined models used for consenting patients for experimental research. However, the emerging LHS model and increasing use of cluster randomisation methods complicate issues regarding the ethical treatment of patients participating in PCTs,6 9–12 because these trials challenge the previous concept that research and clinical care are distinct activities.13 The blending of clinical care and research within clinical practice presents new challenges to investigators and institutional review boards (IRB) regarding which activities should be subject to human subject research regulations and what informed consent requirements are needed to protect the autonomy of research participants.14 15 This article details a novel approach by the Wake Forest University Health Sciences IRB to address the challenges of blending clinical care and research in a PCT. Our approach to analysing and satisfying the ethical obligations to participants in a PCT may be useful for informing ethical practice within the context of pragmatic research and LHS.
Background
The Comprehensive Post-Acute Stroke Services (COMPASS) trial is evaluating the comparative effectiveness of a patient-centred, comprehensive postacute stroke intervention versus usual care.16–18 In North Carolina, 41 hospitals (40 randomised sites plus the lead site already using the COMPASS model) were assigned either to adopt and implement the COMPASS poststroke care model for all eligible patients, or continue usual standard of care (control group). The hospitals were chosen to ensure diversity in stroke patient volumes, geographic locations (ie, rural vs urban) and primary stroke centre certification status. Hospital leadership, research and legal officials, and the neurology departments served as gatekeepers. At each selected site, the gatekeepers provided permission to engage the facility in the research study, but did not provide informed consent for the patient participants. Wake Forest School of Medicine is the lead site for the study.
All patients aged 18 years or older discharged home from a participating COMPASS study hospital with a diagnosis of ischaemic or haemorrhagic stroke or transient ischaemic attack were eligible to participate. Only patients discharged directly home from the hospital were eligible. Patients discharged elsewhere (to a skilled nursing facility and/or nursing home) were more likely to have severe impairments from the stroke and were not included. Those with a diagnosis of subdural or aneurysmal subarachnoid haemorrhage or those who speak neither English nor Spanish were also excluded. These criteria help protect patients without the capacity (cognitive and/or verbal) to give consent from being enrolled in the study. In addition, telephone interviewers are trained to identify individuals who have cognitive impairments. The telephone screener script contains questions modified from the Telephone Interview for Cognitive Status. If the participant is unable to answer these questions correctly, they are excluded from the study or a legally authorised representative consents on their behalf. Approximately 6000 participants are expected to be enrolled in the study.
The intervention
The comprehensive postacute stroke care intervention consists of telephone call from a study team member 2 days after discharge to ascertain if further medical assistance is needed and to schedule any necessary follow-up with the appropriate healthcare provider. At 7–14 days after discharge, an advanced practice provider evaluates the patient using the poststroke functional assessment tool and develops an individualised care plan. At 30 and 60 days after discharge, the participants are called to ask about their care plan and any challenges they are facing. Three letters providing educational material from the American Stroke Association and information about an upcoming 90-day telephone survey are provided. Finally, at 90 days participants receive a telephone survey to assess overall health. Participants at control group sites receive standard treatment, the three letters with educational material and information on the 90-day telephone survey, and the telephone survey to assess overall health at 90 days.
It is important to point out that all of the research activities in COMPASS meet the definition of minimal risk. US regulation defines minimal risk human research as those in which ‘the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.’19 The records review, telephone call, physical examination and survey are all common occurrences outside of the research context and the risks of these experiences are generally not considered greater than those of daily life. The study data were comprised only information gathered from the records, those from the clinical examination and postdischarge telephone call performed in the intervention arm, and the information gathered during the survey call.
Although the research was minimal risk, the obligation to inform individuals and support autonomous decision-making was not lessened. The question of how to provide informed consent was raised early in development of the protocol. Discussions involving the IRB and the study team were invaluable in forging a model of notification and consent to address the ethical obligations to those seeking treatment at a participating hospital, without creating unreasonable burden for the study team or treating clinicians at the sites. This report describes our experience in addressing these concerns within COMPASS.
Assessing ethical obligations related to informed consent
It has been argued that in the context of an LHS, the obligation to respect autonomy in research may differ from that in more traditional research settings. This is because the primary patient goal within an LHS is still a positive health outcome and the minor differences that may be introduced into their care by comparative research are not of importance to patients. This perspective is associated with a proposal for an entirely new ethical framework for the LHS, including the concept of patients’ moral obligation to participate in research.10 In addition, some argue that because low comprehension of informed consent materials is well documented, informed consent is likely not an effective foil against exploitation. Therefore, reliance solely on the IRB review process makes more sense as a primary safeguard.20 However, that would be a substantial departure from the view posited by the Belmont Report, which states that research differs from clinical care because the aims of research are to test hypotheses rather than to focus solely on the well-being of the patient. Individuals should be informed and offered a choice regarding participation in research.4 The elements of informed consent required by US regulations are a statement explaining that the study is research; the purpose of the study; the expected duration of participation; information about the procedures; identification and description of foreseeable risks and benefits; alternatives to participation; a statement about the degree of confidentiality to be expected; contact information; and a statement that participation is voluntary and may be discontinued without penalties.
Recent literature indicates that the public want to be informed and asked their permission to be enrolled in research21 studies; this desire is greater when personal information is collected or sharing of medical information is involved.2 22 23 Our recent national survey supports the argument that the public values autonomy, even in low-risk research.21 The results are not surprising, given previous claims that consent is one of the key components in ethical human research.24 Because clinical research depends on willing participants, meeting the expectations of the public with respect to ethical treatment of participants is important.25 If investigators lose the trust of the public by1 failing to engage them in decision-making,2 failing to disclose that research is occurring, or3 conveying that researchers care only about the data, chances for clinical advancement are damaged far more than if the study had never been done. Trust develops when individuals are confident that others in positions of authority want to treat them with the highest degree of integrity, respect and goodwill.26 Thus, in developing an informed consent design for COMPASS, we were committed to engaging patient and stakeholder perspectives and to finding a way to reach the highest standards, not simply meeting the minimum requirements.
Defining participants
We considered the patients who would receive either standard postacute stroke care or the comprehensive intervention care as participants because they were both the recipients of the research-related care and the individuals about whom identifiable information was collected. We did not consider the care providers at participating sites to be research participants because they were not the subject of the research and identifiable information was not collected about them. To meet our duty to inform participants we carefully considered the ways in which autonomy could be preserved for each patient.
Meeting ethical obligations
To develop an appropriate human research protection plan, the IRB examined each point at which data would be viewed, what data would be recorded, how that data would be captured and used, and the scope and content of the billing claims information to be analysed. At each point, a decision was made regarding the ethical requirement of consent for data acquisition, practicability of written consent, the logistics of obtaining consent and stakeholder input. We also conducted a literature search and considered alternatives to traditional informed consent. We ultimately decided on the use of multiple approaches, including an adaptation of broadcast notification, to notify and inform patients.27
The first thing we determined was that patients did not need to provide research consent to receive either the standard care or the (REDACTED) care model for treatment purposes because in either circumstance they were receiving approved medical care. Those in the standard arm received the usual care offered at their hospital and those at sites providing the COMPASS care model were receiving a level of care that was already deemed appropriate through the Center for Medicare and Medicaid Services value-based care recommendations for caring for complex conditions like stroke.28 However, we believed that patients should be notified about the cluster randomised trial and that they must be provided with an informed consent process for participation in any non-treatment-related study activities at the appropriate time.
To determine who should be notified about their hospitals’ participation in COMPASS, and what that meant for their care, we had to determine who would be affected. It was determined by the IRB that a waiver of the Health Insurance Portability and Accountability Act (HIPAA) authorisation was appropriate for the identification of patients who would be eligible for the study and would therefore need to receive the notification handout about the COMPASS trial. It was not possible to identify eligible participants before the point at which they were cleared for discharge because the inclusion criteria required that patients be discharged to home. So the point of discharge was considered the best opportunity to introduce the study without negatively impacting workflow or causing undue confusion for patients and their families.
Patient and stakeholder perspectives on the process
The study team received input from two former patients with stroke and two caregivers on when and how to notify and consent patients to participate in the study. Their recommendations were incorporated into the process. The former patients with stroke explicitly told the study team that formally consenting eligible patients at discharge was not a good approach, but providing information about the study at that time with consent occurring at a later point would be appropriate. They explained that patients are flooded with information and paperwork at discharge and would not understand what they were signing if consent were offered at that time. Making patients aware of the study at discharge and telling them whether they were receiving care in an intervention or control hospital would be helpful and would provide context for a later consent process.
The patient-stakeholders also reviewed the notification handout being designed by the study team. The patient-stakeholders wanted to make certain that the wording in no way implied that usual care (the control condition) is inferior care. They believed that such a perception could interfere with the patients’ recovery. The patient-stakeholders reviewed several iterations of the handout to make sure that the document was understandable to potential participants with a range of health literacy levels. Our patient-stakeholder partners provided repeated feedback by email with the study team. The IRB maintained its oversight role while also learning from the patient-stakeholders, recognising that the people it protected had ideas that needed to be incorporated into the consent process.
Ultimately, the IRB approved the notification handout (see online supplementary appendix A and B) and it was provided to each eligible patient at discharge to ensure they were informed of their hospitals’ participation in the study and the arm to which their hospital was randomised. At that time patients were told that they would receive a telephone call about 3 months later to request their participation in a voluntary outcomes survey and they received a toll-free telephone number to call if they had questions about the study or wished to be removed from this survey. The notification at discharge served to preserve the autonomy of those identified as potential participants by providing the information they needed to consider regarding the participation of the hospital in the research and the study-only activities that would be offered to them in upcoming weeks.
Supplementary file 1
Methods for consent in COMPASS
To ensure that we fully considered the ethical implications of the possible approaches of consent or waiver, the Wake Forest University Health Sciences IRB consulted a bioethicist, Nancy M. P. King. We all agreed that while IRB oversight of study conduct is itself a critical component in the protection of research participants, IRBs must always ensure that research includes an individual consent process whenever possible. IRBs are also obliged to ensure that the consent process is carried out at a time, and in a way, that maximises the potential for clear understanding of the information provided. Although an IRB cannot guarantee that an individual will fully understand the information, the process must be designed and executed in a manner that gives each individual the opportunity to reach that point of understanding. Thus, the burden of deciding on behalf of individuals, who could practicably make their own choices regarding research participation, would not fall on the IRB.
The IRB-approved consent design is depicted in figure 1. The illustration shows the HIPAA waiver granted for the review of medical records to identify potential participants, determine their eligibility and provide them with the informational handout at discharge. Identifying potential participants without a waiver for review of medical records for diagnosis and discharge information would not have been possible because it would have required getting signed authorisation from all patients or their legally authorised representatives to review these elements in their records. The waiver allowed eligible patients to be informed about the study at the earliest possible time. However, they were not asked to sign any documents at this point since their time to consider the study and the options available to them were limited by the clinical discharge process.

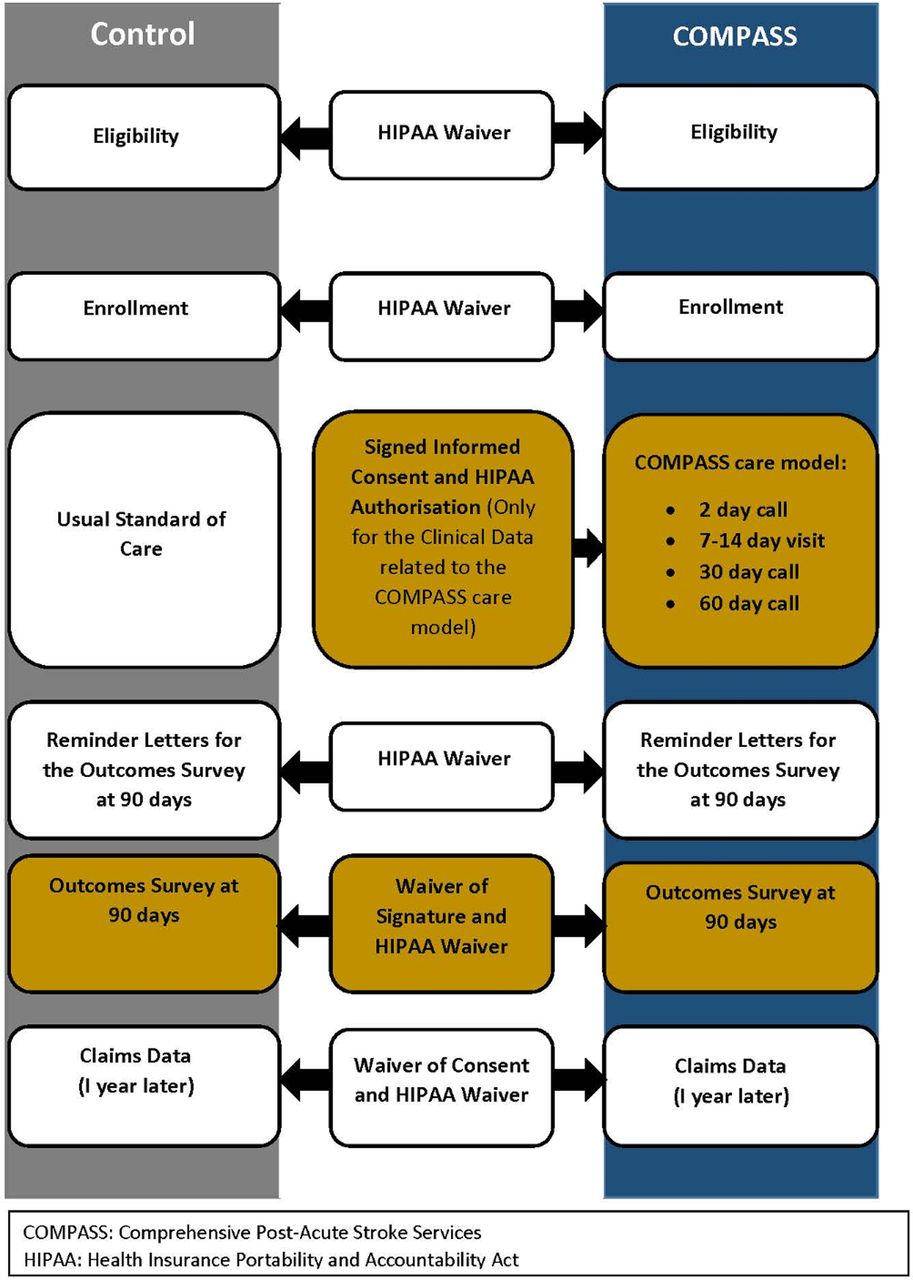{kind=link}
Consent process for the COMPASS trial.
For patients at sites randomised to provide the comprehensive postacute stroke intervention, the 7–14 day postdischarge clinic visit served as the time and location for the consent process. Participants were presented with a written consent form and HIPAA authorisation and these were verbally explained (see online supplementary appendix C). A written signature on the consent form and HIPAA authorisation was obtained from all patients who wished to participate in research-only activities and allow the use of their data in the study. If a patient did not wish to participate in the study, he or she still received the COMPASS follow-up clinical care, but none of their clinical data were used for research purposes.
Patients at sites randomised to standard care did not return for a 7–14 day clinic visit after discharge, so consent for participation in the study was conducted at the next point of active contact; the 90-day postdischarge telephone survey. This telephone conversation provided an opportunity for the study team member to speak individually with the patient. Obtaining signed consent by mail or electronically would have been impracticable given the large number of patients involved. Instead, the IRB approved a waiver of signed consent for those who were treated in the standard care arm. A waiver of signed consent is permissible under US regulations when the research is no more than minimal risk and involves no procedures for which written consent is normally required outside of the research context.19 The study was explained by telephone and verbal consent was obtained from those wishing to participate (see supplementary appendix D). A full waiver of HIPAA was granted for those consenting by telephone because a waiver of signature is not allowed for HIPAA authorisations. Instead, a full waiver is required if signed authorisation cannot be practicably obtained.29
All participants (control and intervention) received communications by mail about a 3-month telephone-based survey. When they were called by the study team, all participants were read a standard informed consent script. Participants verbally consented to continue with the telephone survey. A waiver of consent and HIPAA authorisation was granted for review of billing claims data, which will occur about 1 year after discharge of all previously consented study participants.
Practicality and responsiveness to stakeholder concerns were preserved by obtaining informed consent for all points during the study when data are collected for research-only purposes. The autonomy of individuals was preserved by explaining the study, why it is being conducted, and respecting their choice regarding whether to participate in the additional survey activities. Clinical activities were carried out regardless of participation in the study, but data from those additional clinical components unique to the COMPASS care path are only used for research purposes with signed consent and HIPAA authorisation on file.
The approach we developed to notify and consent participants in the COMPASS cluster randomised trial meets all of the recommendations outlined in the Ottawa Statement on the Ethical Design and Conduct of Cluster Randomized Trials as well as US regulatory requirements and the ethical standards put forth in the Belmont Report.1 30 The use of a cluster randomised design was justified to the IRB and accepted as valid. The gatekeepers at each site made the decision regarding site participation, but did not consent on behalf of individual participants. The team worked with the IRB and with community stakeholders to develop practicable and ethical methods of identifying potential participants. We provided them with information about the study at the earliest time possible and consented them at a time when they were able to ask questions, consider the study carefully and make a personal choice as to whether they wish to participate. No participant was denied the care that they would otherwise receive if there were no study. Each participant received either standard care currently performed at control sites or the standard care plus additional visit and follow-up in the intervention arm.
COMPASS only recruited adult participants. No persons meeting the definition of prisoners were to be enrolled and no payment was provided for participation. Because the Belmont Principle of Justice requires that persons not be unfairly excluded from research, pregnant women were not excluded. However, the intervention was not expected to have any effect on the pregnancy. Study team members employed measures to evaluate the cognitive status of each individual prior to enrolling participants. Those unable to consent for themselves were either not enrolled or were enrolled by a legally authorised representative. By taking these steps, we ensured that the 15 recommendations of the Ottawa Statement were fulfilled and that the right to autonomous decision-making was upheld for the patients at each of the participating sites.
Explaining the consent design
Wake Forest University Health Sciences decided to offer central IRB oversight to participating sites to (1) minimise variation in study conduct and consent; (2) offer a streamlined approval process convenient for sites, particularly those with less clinical trial experience; and (3) align with National Institutes of Health policy on multisite studies and the proposed common rule changes that included a central IRB requirement for federally funded clinical trials.31 32 Although the new federal policies were not yet in effect, the IRB saw COMPASS as a case where the central review concept made sense. As research sites were recruited by the COMPASS study team, telephone calls were arranged between the Wake Forest IRB and each site. During these calls, sites were invited to rely on the central IRB, and the innovative consent design was explained. In general, sites and local IRBs were receptive to the design, but they also identified challenges. Those challenges, and how they were resolved, are summarised in table 1.
Challenges and solutions to ensure respect of persons in the CCOMPASS trial
Once IRB approval at Wake Forest was obtained, recruiting other sites was the next challenge. All participating sites were encouraged to use the central IRB; 36 chose to do so and five elected to require local IRB review. Only minor accommodations were allowed, such as small edits to the consent language; these were then adopted by all 41 participating sites. One hospital decided not to participate primarily because their representative did not think they could accommodate the novel approach within the timeline for our study launch.
The Wake Forest IRB called each site to establish if the site had participated in research, if they had a local IRB and if that local IRB was willing to rely on the review of the central IRB. Study team worked with each site to achieve IRB approval. This approval process varied depending on the site. Multiple follow-up calls and written correspondence occurred with sites, as needed.
The COMPASS team and Wake Forest IRB provided training as needed for both general regulatory requirements (eg, Federalwide Assurance registration) and appropriate study conduct. This time-consuming endeavour was essential to ensure that sites had a solid understanding of the study and how to comply with regulatory and ethical standards.
Conclusion
Our experience shows how successful collaboration between the study team, community stakeholders, a central IRB and multiple sites can help ensure that a complex study is carried out in a way consistent with the Belmont principles and regulatory requirements. Approval of PCTs like COMPASS will require flexibility and collaboration from IRBs. Similarly, successful implementation of the LHS model depends on partnership and collaboration from overseeing IRBs. We also recommend involvement of stakeholders—a critical part of any Patient-Centered Outcomes Research Institute-sponsored study—as a study’s components evolve. However, study teams must be realistic about the investment of time and effort these interactions will take.
The flexibility exercised by the Wake Forest IRB ensured that obtaining consent was not overly burdensome to the COMPASS study team or patient participants. Using the discretion made available to IRBs by regulations, it was possible to shape a consent plan tailored to the needs of potential participants without hindering important research.
Thinking through the steps of a research protocol to find the right points at which consent should be addressed helped the study team and IRB become familiar with the study from each other’s perspectives, and lessens the potential for delay at the point of formal review. While we considered the time committed to this process a net gain, this time commitment should not be underestimated. For trials of this size, we recommend an additional full-time employee (FTE) be dedicated to managing the IRB process during a study’s start-up phase. The most important goals are that participants’ rights are respected and the trust of the community is maintained. Sites relying on a central IRB have confidence that the IRB fully understands the study and has thought through ethical issues carefully. Investigators benefit from the input of experienced IRB members who can provide sound advice.
Finally, the team and the IRB kept the patient perspective central throughout the process. This perspective was important to ensure that (A) patients receive the current best practice or better care during the study; (B) patients and their caregivers see that healthcare systems continue to learn how to improve poststroke care; and (C) patients and caregivers’ choices are respected. This common focus also helped to strengthen relationships among the colleagues at each study site.
Acknowledgments
We acknowledge the individuals who contributed to this work Gladys Lundy, Betsy Vetter, and Maria Orsin . We also acknowledge the editorial assistance of Karen Klein, MA, in the Wake Forest Clinical and Translational ScienceInstitute (UL1 TR001420; PI: McClain) .
References
Footnotes
Contributors JEA, RBW, JBM, PD, PM and DO contributed to the planning and conceptual design of the consent process discussed in this article from the perspective of the Institutional Review Board. They worked with the clinical trial investigators to design the innovative consent process. NMPK is a bioethicist who worked with the IRB and investigators to ensure that the ethical issues were fully explored and that the design chosen was acceptable. CB, WR, JH, SJ and SG contributed to the planning and design of the clinical trial itself, as well as the consent process discussed in this paper. CB, WR and JH contributed to the conduct of the clinical trial itself. SJ and SG contributed to the conduct of the study and the data acquisition and analysis. All of the authors contributed to and reviewed the manuscript prior to submission.
Funding This study was funded by the Patient-Centered Outcomes Research Institute, 10.13039/100006093 (Grant No: PCS-1403-14532) and the National Center for Advancing Translational Sciences, 10.13039/100006108 (Grant No: UL1 TR001420).
Competing interests None declared.
Patient consent Obtained
Ethics approval The Wake Forest University Health Sciences IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Other content recommended for you
- Comparative effectiveness of paediatric kidney stone surgery (the PKIDS trial): study protocol for a patient-centred pragmatic clinical trial
- Clinical Research From Proposal to Implementation
- Reconsidering ‘minimal risk’ to expand the repertoire of trials with waiver of informed consent for research
- Does Waiver of Written Informed Consent from the Institutional Review Board Affect Response Rate in a Low-Risk Research Study?
- Informed consent in pragmatic trials: results from a survey of trials published 2014–2019
- A rationale and framework for seeking remote electronic or phone consent approval in endovascular stroke trials – special relevance in the COVID-19 environment and beyond
- Remote monitoring of medication adherence and patient and industry responsibilities in a learning health system
- Deferred consent for the enrolment of neonates in delivery room studies: strengthening the approach
- Randomised comparative effectiveness trial of Pulmonary Embolism Prevention after hiP and kneE Replacement (PEPPER): the PEPPER trial protocol
- Informed consent in cluster randomised trials: a guide for the perplexed