Article Text

Abstract
When may a physician enroll a patient in clinical research? An adequate answer to this question requires clarification of trust-based obligations of the state and the physician-researcher respectively to the patient-subject. The state relies on the voluntarism of patient-subjects to advance the public interest in science. Accordingly, it is obligated to protect the agent-neutral interests of patient-subjects through promulgating standards that secure these interests. Component analysis is the only comprehensive and systematic specification of regulatory standards for benefit-harm evaluation by research ethics committees (RECs). Clinical equipoise, a standard in component analysis, ensures the treatment arms of a randomised control trial are consistent with competent medical care. It thus serves to protect agent-neutral welfare interests of the patient-subject. But REC review occurs prior to enrolment, highlighting the independent responsibility of the physician-researcher to protect the agent-relative welfare interests of the patient-subject. In a novel interpretation of the duty of care, we argue for a “clinical judgment principle” which requires the physician-researcher to exercise judgment in the interests of the patient-subject taking into account evidence on treatments and the patient-subject‘s circumstances.
- RCT, randomised controlled trial
- REC, research ethics committee
Statistics from Altmetric.com
With increasing demand for contemporary medicine to be placed on a firmer scientific foundation, clinical research is enjoying unprecedented expansion. Accordingly, doctors in increasing numbers are participating in clinical trials, as are greater numbers of patients. Hence, it is surprising that the central moral dilemma of the randomised controlled trial (RCT) remains so vexing: when may physicians, consistent with their duty of care to patients, offer the them enrolment in an RCT?1 If reasonable public trust in the ethical integrity of clinical research is to be maintained, a convincing answer to this question must be given. Presently, we explore the dilemma, examine solutions that have failed to galvanise support, and offer a new one.
The dilemma arises because offering patients enrolment in RCTs imperils the doctors’ duty to act in their interests. This is because, in seeming contrast to clinical practice, an activity structured to protect and advance the medical interests of the patient, clinical research necessarily calls for the pursuit of other interests, including the public interest in the production of generalisable knowledge, and the variable private interests of industry, institutions and researchers. The pursuit of these interests inevitably influences the conduct of clinical research materially. To ensure scientifically valid results, treatment in a clinical trial is restricted by design features of the study protocol (eg, randomisation to treatment, blinding of treatment allocation and fixed schedules for treatment provision). Further, patient-subjects typically undergo non-therapeutic procedures to answer the scientific question. In these ways, the demands of science place the conduct of clinical trials in tension with the doctor-researcher’s positive duty of care (hereinafter referred to as duty of care) to the patient-subject.
Hellman and Hellman2 have argued that an unmanageable conflict exists between the interests of the patient-subject and those of others in clinical research. Conducting RCTs, in their view, necessarily violates the rights of patient-subjects to have their interests protected. They view the rights violation as being of such magnitude that RCTs on patients must be abandoned and replaced by non-randomised forms of clinical research. We believe that their position is untenable. RCTs have a pivotal role in the generation and scrutiny of the evidentiary foundation of clinical practice. The results of clinical research are not merely of private and academic interest; they further the public good of high quality medical care. Without clinical research, much of the practice of medicine would remain untested, and medical progress would be stymied. Furthermore, we question whether the tension between the demands of science and the rights of patients results in an insoluble conflict of interest, requiring an either/or solution.
A recent proposal from the US purports to solve the dilemma by denying its existence. Miller and colleagues deny that doctor-researchers operate under a duty of care to patient-subjects. They argue as follows. Clinical practice and clinical research have differing goals. The goal of clinical practice is to provide the patient with optimal care. The goal of research is to produce generalisable knowledge that may benefit future patients and society. They assert that because the goals of clinical practice and clinical research differ, the norms governing each must not overlap.3 Implicit is the suggestion that the physician-researcher ceases to be a physician when conducting research by virtue of the nature of research as an activity. They explicitly deny that the physician-researcher has a positive obligation to act in the interests of the patient-subject when conducting research.4,5 The only duties of the physician-researcher to the patient-subject are to obtain informed consent and avoid exploitation.
The solution given by Miller and colleagues is unsatisfactory. It has already been noted that an implausible form of moral dissociation is required, whereby doctors engaging in research must wilfully ignore the professional obligations they have as doctors.6 Further, the assertion that activities themselves generate norms is counterintuitive and requires argument. Finally, the additional assertion that the norms governing activities with differing ends are non-overlapping is obviously false. Norms prohibiting fraud and murder are universal and thus apply across diverse activities. Worse still, Miller et al’s view is internally inconsistent on this very point. They recognise a duty to obtain informed consent applicable to clinical practice and clinical research. Thus, at least one norm in their schema is overlapping, refuting a central premise of their argument.
Given this, we think physician-researchers will rightly continue to wonder about the implications of their duty of care to patient-subjects when conducting clinical research. When, consistent with this obligation, can a doctor offer a patient enrolment in an RCT? Is approval by the research ethics committee (REC) sufficient? Is there a role for clinical judgement when doctors act as physician-researchers? To consider these questions and the broader dilemma satisfactorily, in our view, requires recognition and elucidation of the independent obligations of the state and the doctor-researcher to protect the interests of patient-subjects. These obligations are understood to be derived from the trust-based relationship between the patient-subject and the state and the doctor-researcher, respectively.
OBLIGATIONS OF THE STATE
Clinical research is the source of a critical public benefit: it aims to place medical practice on a foundation of scientific evidence. Without the voluntary participation of patient-subjects, clinical research cannot proceed and the public will be deprived of an important good. Patient-subjects reasonably trust that the state will protect them in exchange for their contribution to the public good of science. As a result of the trust shown by patient-subjects, and the public benefit derived from their participation in clinical research, the state is morally obliged to exercise its powers to protect their interests. The state fulfils its trust-based obligation to protect the interests of patient-subjects through promulgating regulations with adequate substantive and procedural safeguards for subjects and by ensuring adequate enforcement of these regulations. National regulations and guidelines define standards for scientific and ethical acceptability of clinical research. RECs and national oversight authorities provide an arms length review of clinical research to ensure its compliance with national standards. The REC, therefore, is best understood as an arm of the state that ensures protection of the liberty and welfare of citizens who give of themselves to further medical knowledge.
Clinical research is reviewed prospectively by the REC. Before potential patient-subjects may be approached regarding study participation, an REC review ensures that protocols meet general ethical and scientific standards.7 In carrying out this prospective review, RECs scrutinise several key aspects of the research protocol. The study must be scientifically valid—that is, the methods must be appropriate to answer the study question. The study question posed must be of sufficient value to science and society to justify non-therapeutic risks to patient-subjects. Procedures to enrol patient-subjects in the study must be fair and neither exclude those who may benefit from participation nor enrol those who may be unduly susceptible to harm. The benefits and harms of study participation for patient-subjects must stand in reasonable relation. Adequate procedures must be in place to secure valid informed consent. Finally, sufficient safeguards must be in place to protect the confidentiality of private health information.
In making judgements pertaining to liberty and welfare, the REC does not take into account the agent-relative interests of patient-subjects. Review of clinical research by RECs is prospective and, as such, particular patient-subjects and their particular interests are not yet in view. Rather, REC review is aimed at protecting the agent-neutral interests of patient-subjects. It ensures that patient-subjects are not exposed needlessly to the risks of a study that cannot answer the scientific question posed, or one that deals with a trivial question. It checks procedures for patient-subject selection, but does not check that each enrolled patient-subject is in fact eligible for study participation. It assesses the benefits and harms of the study in the light of existing data on the treatments and characteristics of the study population, not in light of the medical history of particular patient-subjects. The REC scrutinises consent procedures and documents, not processes of obtaining informed consent from particular patient-subjects. It requires that adequate procedures are in place for protecting the privacy and confidentiality of the personal information of patient-subjects, but does not supervise the handling of information of particular patient-subjects.
Of all of the norms used by the REC in protecting the agent-neutral interests of patient-subjects, the least understood are the substantive and procedural norms through which RECs ensure that benefits and harms of study participation stand in reasonable relation. Existing regulatory norms relating to the evaluation of research benefits and harms are typically vague and disconnected. Interpretations of key concepts vary widely.8 RECs are thus left to make intuitive assessments on the acceptability of benefits and harms of the research, contributing to inconsistent decision-making processes and results. Elsewhere, we have worked to remedy this problem by providing a structured set of norms and procedures for benefit–harm evaluation by the REC (box 1).
The role of the REC is to fulfil the state’s obligation to protect the agent-neutral interests of patient-subjects. It does so, partly, through ensuring that the ethical requirements of component analysis, including clinical equipoise, are satisfied.
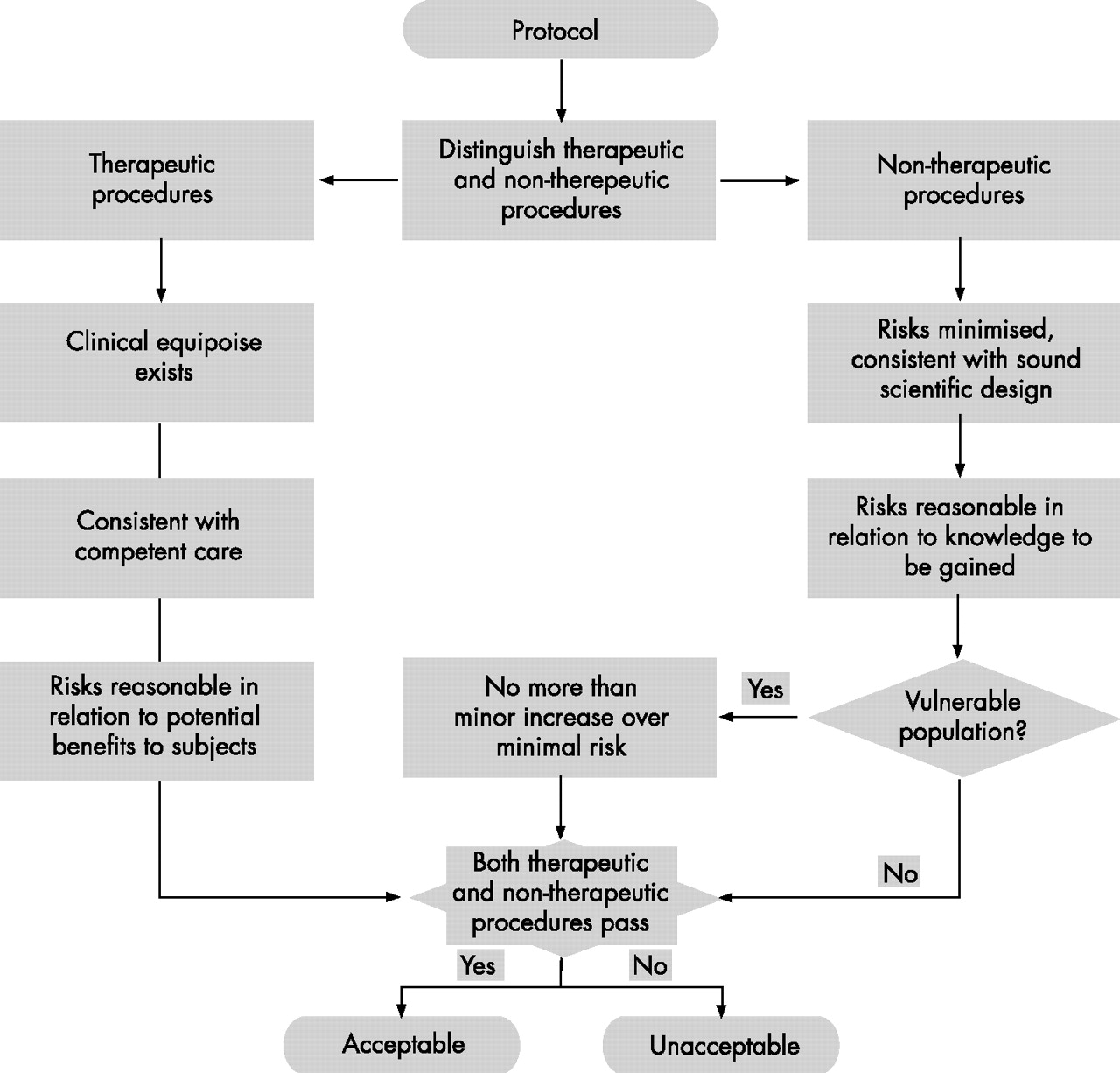
We have named our approach component analysis, for it rests on the insight that clinical research often contains a mixture of study procedures.9 Some procedures in clinical trials are administered with therapeutic warrant (“therapeutic procedures”), whereas others in the absence of therapeutic warrant are administered purely to answer the scientific question at hand (“non-therapeutic procedures”). The distinction in terms of the presence of therapeutic warrant is morally relevant. Only procedures administered with therapeutic warrant can reasonably be said to advance the welfare interests of patient-subjects in receiving treatment. The benefits and harms associated with therapeutic procedures may therefore be justified through a comparative assessment of the benefits and harms of available treatments. Non-therapeutic procedures cannot reasonably be said to advance the welfare interests of patient-subjects. The risks associated with non-therapeutic procedures must therefore be justified by standards that weigh the welfare interests of patient-subjects in protection from harms of research against the interests of the public and others in the benefits of research. The moral condition imposed on the use of therapeutic procedures is intended to protect their welfare interests in receiving competent medical treatment. By contrast, moral conditions imposed on the use of non-therapeutic procedures are intended to secure their welfare interests in protection from unreasonable risks solely in the interests of others.
On the basis of this distinction, component analysis provides separate substantive moral standards for REC evaluation of the benefits and harms of therapeutic and non-therapeutic procedures (fig 1). When protocols provide for both therapeutic and non-therapeutic procedures, an REC may deem that a study poses an acceptable balance of benefits and harms only if both sets of moral standards are fulfilled. Obviously, when protocols provide only for non-therapeutic procedures (eg, interview studies or phase-I drug trials in healthy people), this determination is contingent only on satisfaction of standards applicable to non-therapeutic procedures.
{kind=link}
Ethical analysis of benefits and harms in research by the research ethics committee.9
Therapeutic procedures must satisfy the clinical equipoise requirement (fig 1).10 That is, to approve randomisation of subjects to the therapeutic procedures to be studied in the trial, the REC must find that there is a state of honest, professional disagreement in the community of expert practitioners as to the preferred treatment. The prospective and general nature of REC review, combined with the inherently social nature of the establishment of treatment standards, make it appropriate for the REC to evaluate therapeutic procedures in the light of the state of community opinion on the comparative merits of available treatments. In ensuring that the expert community is at odds regarding the comparative merits of treatments available to patient-subjects, the REC ensures that they will not be asked to accept substandard treatment to participate in clinical research. Procedurally, to make this determination, the REC does not survey practitioners. Rather, it scrutinises the justification for the study, the relevant literature and, where needed, the opinion of impartial clinical experts. Therapeutic procedures are acceptable when the REC judges that, were the evidence supporting the various therapeutic procedures widely known, expert clinicians would disagree as to the preferred treatment.
As non-therapeutic procedures are not administered with therapeutic warrant, differing moral standards apply to their evaluation. In general, risks associated with non-therapeutic procedures must be minimised, consistent with sound scientific design, and must be reasonable in relation to the knowledge to be gained (fig 1). Procedurally, the REC ensures that all proposed procedures are actually required to meet the scientific ends of the study. At times, non-therapeutic risks may be minimised by piggybacking on routine clinical procedures—for example, extra blood for research purposes may be taken at the time of a clinically indicated blood draw, thereby saving the patient-subject an additional venepuncture. Whether non-therapeutic risks are reasonable in relation to the knowledge to be gained requires a judgement that draws on both scientific expertise on the REC and the opinion of community representatives on the social value of the scientific ends pursued. When research is conducted on a vulnerable population, such as children, an additional moral standard applies, whereby risks associated with non-therapeutic procedures are limited to a minor increase above the minimal risk. Minimal risks are potential harms unthinkingly assumed as a matter of routine in daily life.11 Procedurally, the REC must determine that the risks of non-therapeutic procedures are in fact risks of daily life (eg, physical examination) or sufficiently equivalent to be considered roughly interchangeable (eg, ultrasound). Whether the daily lives referred to by the minimal risk standard ought to be those of healthy or sick people remains controversial.12,13
Component analysis provides RECs with a comprehensive and systematic framework for evaluating the acceptability of benefits and harms in research. It thereby offers the prospect of improving the quality and consistency of REC review, enabling RECs to better meet the trust patient-subjects repose in the state. However, it is important that we realise the limitations of REC approval. As with its enforcement of other norms, so in its enforcement of the norms related to benefits and harms in component analysis, REC approval signifies only that a protocol meets general standards mandated by the state. The scope of REC review is limited to protection of the agent-neutral interests of patient-subjects, and hence the moral and legal significance of REC approval is circumscribed. REC approval does not entail the moral or legal acceptability of enrolling particular patient-subjects in research, nor does it entail the acceptability of their continued participation in the study, as these acts engage the agent-relative interests of patient-subjects. It is the doctor-researcher who retains the independent moral and legal obligation to protect the agent-relative interests of the patient-subject in clinical research.
OBLIGATIONS OF THE DOCTOR-RESEARCHER
Miller et al’s arguments deny that the doctor-researcher is bound by a duty of care to protect the agent-relative welfare interests of the patient-subject in clinical research. Their position is without persuasive moral or legal foundation. Their account of the obligations of the doctor-researcher to the patient-subject has the hallmarks of contractualism. Most notable is the overriding emphasis on informed consent and the negative obligation to avoid exploitation. Although the norms of contractual relationships have an essential role in the social and economic order, they are unsuited to the relationship between a physician-researcher and patient-subject. We leave open the question of whether the contractual model is an appropriate one for relationships between healthy participants and researchers.
It has long been recognised that trust forms the foundation of the relationship between a doctor and a patient. The central normative importance of trust to the relationship has been recognised by doctors and protected by fiduciary law for several important reasons. First and foremost, the doctor–patient relationship is rooted in the frailty of the human condition. People are vulnerable to sickness and disease and are often made vulnerable by illness when entering into relationships with doctors. Furthermore, the state vests exclusively in doctors the authority to receive, diagnose and treat patients. By virtue of this grant of exclusive professional license, coupled with disparities in knowledge and power, patients must depend on doctors for the beneficial exercise of clinical judgement in maintaining and improving their health.
Coming to a relationship often prefigured by circumstantial inequality of power and dependence, patients are doubly vulnerable on account of the structural inequality of power and dependence generated by the very act of entrusting power over one person’s interests to another—namely, the doctor. Circumstantial and structural inequality of power and dependence give rise to a heightened vulnerability to exploitation on the part of the patient-subject. In short, in entrusting power to doctors, patients necessarily face the risk that this power will be misused to their detriment by doctors in pursuit of their own interests or those of others. Precisely because of the vulnerability that prefigures and is generated by it, the relationship between a doctor and a patient is closely monitored and policed by the profession and the state.
The trust model is the appropriate one for the relationship between doctor-researcher and patient-subject. The relationship bears all the hallmarks of a trust relationship. The patient-subject cannot reasonably be expected to act as a shrewd, independent party to a contract, enjoying an arms-length relationship with the doctor-researcher in which both fend for their own interests. Many patients considering participation in research continue to be made vulnerable by illness. They usually agree to participate in research hoping thereby to receive improved treatment. Through consent, the doctor-researcher is authorised to exercise most, if not all, of the discretionary powers enjoyed by the doctor, including powers of diagnosis and treatment. Similar to other trust relationships, the relationship between physician-research and patient-subject is characterised by circumstantial and structural inequality of power and dependence relating to authorisation of the exercise of power. By virtue of their illness and their desire to overcome it, patient-subjects authorise doctor-researchers to exercise discretionary powers, and trust and rely on them to do so in their interests to the greatest extent possible given the inherent demands of research design.
If the relationship between a doctor-researcher and a patient-subject is properly theorised as one of trust, what obligations accrue thereby to the doctor-researcher? We suggest that trust relationships of this kind—that is, in which one party entrusts another with power over significant practical interests—give rise to a range of obligations reflecting the structure of the relationship. They include a duty of loyalty and a duty of care. We do not explore all trust-based obligations presently, as we are here interested only in the manner in which one of them—the duty of care—is to be specified in consideration of a particular trust relationship—namely, that between physician-researcher and patient-subject.
The specification of the duty of care will vary among relationships depending on the scope of power wielded by the stronger party and how the interests of the weaker party are defined. Patient-subjects in clinical research by definition are ill and have an interest in receiving competent medical care. Doctor-researchers by virtue of their professional licence have the power to provide such care. The duty of care under which doctor-researchers operate may thus be specified as follows. The doctor-researcher is trusted to exercise clinical judgement in protecting the medical interests of the patient-subject and has an obligation to act in a manner that meets this trust.
This simple statement of the duty of care belies considerable difficulty and disagreement over its precise specification. Just how are doctor-researchers to meet their obligation to exercise judgement in the interests of patient-subjects, given the restrictions trial design places on treatment? Peto14 has provided a popular specification of this obligation in the form of the uncertainty principle. According to it, “[i]f … [physician-researchers] think, whether for a wise or silly reason, that they know the answer before the trial starts, they should not enter any patients [in the RCT]….”.14 Put the other way round, the doctor-researcher may offer a patient enrolment in as to RCT when the doctor-researcher is genuinely uncertain as to the preferred treatment for the patient. If the physician-researcher has a preference for one treatment over another, she may not offer the patient enrolment in the trial. Whether the doctor-researcher’s preference is based on the medical literature, opinion of colleagues, personal experience or a hunch is irrelevant.
Freedman10 emphasised intractable problems with moral standards, which, like the uncertainty principle, rest the ethics of RCTs solely on the judgement of the doctor-researcher and epistemically validate judgements based on weak or no evidence. He argued that all such standards are fragile in that they are unduly sensitive to the vagaries of intuition and opinion. Furthermore, they fail to discriminate between forms and degrees of evidence supporting the judgement of doctor-researchers. Finally, they are insufficiently precise to the extent that they lack specification of the degree of uncertainty required for their satisfaction. Freedman’s alternative is clinical equipoise. According to clinical equipoise, an RCT is permissible if at the outset of the trial there is a state of honest, professional disagreement in the community of expert clinicians as to the preferred treatment. Although Freedman’s critique of the uncertainty principle and similar standards is compelling, clinical equipoise does not adequately specify the doctor-researcher’s duty of care to the patient-subject.
Clinical equipoise forms an integral part of component analysis, providing RECs with a clear standard for the evaluation of benefits and harms of therapeutic procedures in clinical research. However, clinical equipoise directs attention only to the state of expert opinion on the comparative therapeutic merits of treatments for proposed study populations. It is thus limited to protecting the agent-neutral welfare interests of patient-subjects. Clinical equipoise does not contemplate the particular circumstances of individual patient-subjects. Therefore, it is not, and indeed cannot be, considered to be an adequate specification of the duty of care of doctor-researchers, because they are bound to protect the agent-relative welfare interests of the patient-subjects (box 2).
The responsibility of the doctor-researcher is to protect the agent-relative welfare interests of the patient-subject. Knowing that a study has received REC approval, the physician-researcher may only enrol patient-subjects in research in accord with the clinical judgement principle.
A novel specification of the doctor-researcher’s duty of care to the patient-subject is required that avoids the pitfalls of Peto’s approach while keeping the individual patient-subject in view. Given the requirement for review and approval of RCTs by RECs, this specification should also be temporally situated. The doctor-researcher may approach a patient for enrollment in an RCT only after REC approval is obtained. As such, when it is based on appropriate implementation of component analysis, REC approval provides the physician-researcher with a reasonable basis for believing that the enrolment of patients in the RCT is provisionally consistent with their duty of care to the patients. This is because clinical equipoise protects the agent-neutral welfare interests of patient-subjects in receiving competent care. Nevertheless, the use of clinical judgement by the doctor-researcher is essential to ensuring that the agent-relative welfare interests of the patient-subject are protected. Accordingly, to satisfy their duty of care to the patient-subjects, the conduct of the doctor-researchers is subject to what we call the clinical judgement principle. Under the clinical judgement principle, knowing that an RCT has been approved by an REC under component analysis, the physician may offer patients enrolment in a trial unless (1) they believe that it would be medically irresponsible to do so and (2) this belief is supported by evidence that ought to be convincing to colleagues.
Physician-researchers meet their duty of care through making expert judgements, taking account of evidence on treatment alternatives and the circumstances of the patient-subject. In fulfilling the duty, the doctor-researcher will decline to offer enrolment to a patient who meets eligibility criteria, but whose medical history suggests that participation may be unduly harmful. The doctor-researcher must also recommend that a patient-subject withdraw from an RCT regardless of protocol requirements in the case of an unexpected adverse event that would, in clinical practice, require cessation of treatment or adoption of an alternate course of treatment.
The clinical judgement principle has at least four advantages over previous specifications of the doctor-researcher’s duty of care. Firstly, it recognises and defines the place for the clinical judgement of the doctor-researcher in protecting the agent-relative welfare interests of the patient-subject. Clinical equipoise does not leave room for such judgement. Secondly, it situates the exercise of clinical judgement temporally, recognising that the doctor-researcher may not approach a patient for enrolment in an RCT until after the study has been approved by an REC. The uncertainty principle does not speak to REC approval or the relationship between judgements made by the REC and doctor-researcher. Thirdly, it provides a reasonable epistemic basis for doctor-researcher judgement, avoiding the undue restrictiveness of clinical equipoise (ie, only RCT results count as evidence) and the undue laxity of the uncertainty principle (ie, anything counts as evidence). The clinical judgement principle allows judgements to be based on many sources of evidence, including the doctor’s experience, the patient-subject’s history or novel findings in the literature, provided that such evidence ought to be deemed convincing by colleagues. Finally, the clinical judgement principle specifies a duty of care that is, in turn, firmly rooted in the moral and legal theory of trust relationships. Clinical equipoise and other principles offered as specification of the duty of care have been vulnerable to criticism because of a failure to clearly articulate the foundations for the duty in moral and legal theory.
CONCLUSION
The central dilemma of the RCT—namely, when may physicians, consistent with their duty to act in the interests of patients, offer patients enrolment in an RCT—has long resisted a convincing solution. We found that the most intuitively appealing solutions deal with different aspects of the problem. Clinical equipoise provides clear guidance for RECs, while leaving no room for the clinical judgement of doctor-researchers. The uncertainty principle appropriately acknowledges the irreducible place for the doctor-researcher’s judgement, while failing to explicate the protective obligation of the state. For years, these solutions have been viewed as mutually exclusive, each attracting its gainsayers.
Our solution begins with the novel recognition that the state and doctor-researcher have independent trust-based obligations to protect the interests of patient-subjects. As the state relies on the voluntarism of patient-subjects to further the public good of medical knowledge, it operates under an obligation to protect the interests of patient-subjects. The REC fulfils this obligation on behalf of the state when it reviews research prospectively, ensuring that study treatments are consistent with competent medical care as required by the clinical equipoise standard in component analysis. The scope of REC review, however, has inherent limits and attention to these limits emphasises the doctor-researcher’s independent obligation to protect the interests of the patient-subject. We think that this obligation is best understood as a duty of care grounded in the trust-based nature of the relationship between a doctor-researcher and a patient-subject. Like all trust-based duties of care, it stands in need of specification. We provide such specification in the clinical judgement principle.
In contrast with previous solutions to the central dilemma of the RCT, our solution situates it within a normative theory of trust through which we elucidate and specify the independent obligations of the state and physician-researcher. It also provides pragmatic guidance. Firstly, may doctors participate in clinical trials when they have a preference for one study treatment? Assuming component analysis is correctly applied, REC review ensures that the study treatments are consistent with competent medical care. Although the participating doctors may have a preference for one treatment, other equally competent doctors may hold differing views. Physician-researchers ought to set aside their preferences and recognise that only a well-conducted RCT can provide the evidence necessary to determine which treatment is to be preferred.
Secondly, what is the role for the doctor-researcher’s clinical judgement? We have argued that the doctor-researcher has an obligation to protect the agent-relative welfare interests of the patient-subject. REC review entails a population-level judgement that the various study treatments are acceptable. It remains for the physician-researcher to meet her obligations to the patient-subject through the exercise of clinical judgement that takes into account the circumstances of the patient-subject. If the doctor-researcher judges study participation to be medically irresponsible based on of evidence that ought to be convincing to colleagues, she is obliged to decline to offer enrolment or to withdraw the patient-subject from the study.
Acknowledgments
We thank the reviewers of the Journal of Medical Ethics for constructive suggestions for improvement.
REFERENCES
Footnotes
-
Funding: PBM was supported by a doctoral fellowship from the Social Sciences and Humanities Research Council of Canada. CW was supported by a Canada Research Chair (Tier I) and a Canadian Institutes of Health Research operating grant.
-
Competing interests: None.
Other content recommended for you
- Refuting the net risks test: a response to Wendler and Miller’s “Assessing research risks systematically”
- Thinking clearly about the FIRST trial: addressing ethical challenges in cluster randomised trials of policy interventions involving health providers
- Assessing research risks systematically: the net risks test
- Health policy and systems research: towards a better understanding and review of ethical issues
- Intervening in clinical research to prevent the onset of psychoses: conflicts and obligations
- Should patients be allowed to veto their participation in clinical research?
- Do we really know how many clinical trials are conducted ethically? Why research ethics committee review practices need to be strengthened and initial steps we could take to strengthen them
- Beyond informed consent: the therapeutic misconception and trust
- Equipoise, standard of care and consent: responding to the authorisation of new COVID-19 treatments in randomised controlled trials
- Acceptability of donor funding for clinical trials in the UK: a qualitative empirical ethics study using focus groups to elicit the views of research patient public involvement group members, research ethics committee chairs and clinical researchers