Article Text

Abstract
Background Germline pathogenic variants in SDHB/SDHC/SDHD are the most frequent causes of inherited phaeochromocytomas/paragangliomas. Insufficient information regarding penetrance and phenotypic variability hinders optimum management of mutation carriers. We estimate penetrance for symptomatic tumours and elucidate genotype–phenotype correlations in a large cohort of SDHB/SDHC/SDHD mutation carriers.
Methods A retrospective survey of 1832 individuals referred for genetic testing due to a personal or family history of phaeochromocytoma/paraganglioma. 876 patients (401 previously reported) had a germline mutation in SDHB/SDHC/SDHD (n=673/43/160). Tumour risks were correlated with in silico structural prediction analyses.
Results Tumour risks analysis provided novel penetrance estimates and genotype–phenotype correlations. In addition to tumour type susceptibility differences for individual genes, we confirmed that the SDHD:p.Pro81Leu mutation has a distinct phenotype and identified increased age-related tumour risks with highly destabilising SDHB missense mutations. By Kaplan-Meier analysis, the penetrance (cumulative risk of clinically apparent tumours) in SDHB and (paternally inherited) SDHD mutation-positive non-probands (n=371/67 with detailed clinical information) by age 60 years was 21.8% (95% CI 15.2% to 27.9%) and 43.2% (95% CI 25.4% to 56.7%), respectively. Risk of malignant disease at age 60 years in non-proband SDHB mutation carriers was 4.2%(95% CI 1.1% to 7.2%). With retrospective cohort analysis to adjust for ascertainment, cumulative tumour risks for SDHB mutation carriers at ages 60 years and 80 years were 23.9% (95% CI 20.9% to 27.4%) and 30.6% (95% CI 26.8% to 34.7%).
Conclusions Overall risks of clinically apparent tumours for SDHB mutation carriers are substantially lower than initially estimated and will improve counselling of affected families. Specific genotype–tumour risk associations provides a basis for novel investigative strategies into succinate dehydrogenase-related mechanisms of tumourigenesis and the development of personalised management for SDHB/SDHC/SDHD mutation carriers.
- cancer: endocrine
- genetics
- molecular genetics
- oncology
- genetic epidemiology
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
The succinate dehydrogenase (SDH) enzyme complex comprises four subunits (A–D), localises to the inner mitochondrial membrane and catalyses the oxidation of succinate during the Krebs cycle. Three subunit genes (SDHB/SDHC/SDHD) are among the most important susceptibility genes for the neuroendocrine tumours head and neck paraganglioma (HNPGL) and phaeochromocytoma and paraganglioma (PPGL).1 2 HNPGLs are derived from parasympathetic-derived ganglia, whereas PPGLs are derived from sympathetic ganglia and usually secrete catecholamines. HNPGL and PPGL have a strong genetic basis, with approximately 40% being associated with a germline mutation in one of at least 15 genes.3
Since the identification of the role of SDHB/SDHC/SDHD in inherited PPGL/HNPGL at the start of this century,4–6 testing for germline mutations in these genes has become part of standard medical practice. As testing became more widespread, it was revealed that mutations in SDHC and SDHD were associated with a higher risk of HNPGL than PPGL, with the reverse for SDHB.7–10 SDHB mutations are associated with a higher risk of malignancy and renal carcinoma than mutations in other subunits.7 10 11 Initially, the penetrance of SDHB mutations was estimated at ~70%–80%, and intensive surveillance programmes were recommended, but more recent estimates suggest the tumour risk is <50% in non-probands.7 12–14 The combination of malignancy risk with incomplete penetrance makes designing an optimal surveillance programme for asymptomatic SDHB mutation carriers difficult. More accurate predictions of life-time tumour risks and identification of subgroups with higher or lower risks would enable the development of personalised management and surveillance strategies.
Previously we reported the mutation spectrum and genotype–phenotype correlations in 358 patients with germline mutations in SDHB and SDHD.10 Here we report the results of analysis on an expanded cohort of germline SDHB/SDHC/SDHD mutation carriers and stratify missense mutations according to predicted effects on structure and function. We provide more accurate estimates of tumour-specific risks, confirm the mutation-specific phenotype of the SDHD p.Pro81Leu mutation and identify a novel candidate genotype–phenotype association of SDHB missense mutations with effects on SDHB protein stability.
Materials and methods
Patient cohort
The study sample consisted of men and women referred for SDHB/SDHC/SDHD mutation analysis in National Health Service diagnostic laboratories for a personal or family history of PPGL/HNPGL. Carriers of pathogenic mutations in other genes were excluded from this study. Clinical information was collected via a standard pro forma (see online supplementary figure 1), or clinical records, for research studies or a service evaluation study. Non-probands (those tested after a SDHB/SDHC/SDHD mutation was detected in their relative) were included except those with maternally inherited SDHD mutations who, because of a parent of origin effect, will have a tumour penetrance more similar to that of the general population.15 One proband, described previously,16 had a maternally inherited SDHD mutation. Affected individuals were diagnosed by routine clinical investigations. The work described was performed in accordance with the Declaration of Helsinki. All participants gave informed consent for genetic testing. The service evaluation study was approved by Birmingham Women’s Hospital R&D Office. A subgroup of patients (n=401) have been described previously6 10 14 17–19 with 358 included in Ricketts et al.10 We refer to patients as having ‘detailed clinical information’ if we have received a clinical information proforma or have access to their records to ascertain the presence or absence, and age of onset, of tumours. Patients without detailed clinical information were those for whom we had the genetic test report but insufficient extra information to include them in the penetrance analyses (they were censored at age zero). Clinical information was collected at the time of genetic testing and prior to baseline biochemical and imaging screening of asymptomatic gene carriers.
Supplementary file 1
Molecular genetic analysis
SDHB/SDHC/SDHD mutations were detected by Sanger sequencing, next-generation sequencing assay20 or multiplex ligation-dependent probe amplification (MLPA) analysis (SALSA MLPA Kit P226; MRC-Holland, Amsterdam, The Netherlands) (details available on request).
SDHB/SDHC/SDHD sequence variants were classified as pathogenic/benign/variants of uncertain significance (VUS) by the reporting diagnostic laboratory. As methods of variant classification were not uniform between laboratories, all classifications were compared with the ClinVar classification,21 where available. Where there was a disagreement between the local laboratory classification and the ClinVar classification, the variant was described as VUS. This method was chosen to minimise the chance of underestimating penetrance of tumours because of inclusion of families without a true pathogenic variant. Tumour risks and genotype–phenotype correlations were calculated in individuals with variants considered to be pathogenic or likely pathogenic by the diagnostic laboratory (65 probands with a VUS were excluded from penetrance calculations).
In silico protein structure analysis
The DUET and mCSM-PPI scoring systems22–24 were used to predict the structural consequences of missense mutations on protein stability and protein–protein affinity, respectively, using the models of SDHB, SDHD and the succinate complex (see supplementary information).
Statistical analysis
Tumour risks were estimated by Kaplan-Meier analysis (SDHB/SDHD/SDHC). All statistical tests were performed using the programming language R unless otherwise stated.25 The ‘survfit’ function from the survival package was used for Kaplan-Meier survival analysis and penetrance calculations. The log-rank test was used to compare survival distributions between cohorts of different genotypes. To account for the non-random ascertainment of study participants with respect to their disease status, separate penetrance estimation analyses were carried out, in which we modelled the retrospective likelihood of the observed mutation status conditional on the disease phenotypes26 (online supplementary material). The analysis was carried out in the pedigree analysis software MENDEL.27 The retrospective cohort analysis was not used for SDHD, because it does not address the fact that the ascertainment of SDHD mutation carriers considered the parental origin of the mutation. The 0.05 level of significance was used for all tests, with Bonferroni correction for multiple comparisons where stated. All tests were two sided.
Results
Spectrum of SDHB/SDHC/SDHD mutations
Of 1832 patients referred for genetic testing due to a personal or family history of PPLG/HNPGL, 1093 were probands and 1227 had detailed clinical information available. Eight hundred seventy-six had a mutation in either SDHB (n=673, probands=275), SDHC (n=43, probands=26) or SDHD (n=160, probands=90) (see online supplementary figure 2).
Copy number abnormalities (CNAs)
Forty-five probands had MLPA-detected single/multiple exon deletions or duplications in SDHB (n=36), SDHC (n=6) or SDHD (n=3) (see online supplementary table 1). Three of 36 SDHB probands with a CNA had a single or multiexon duplication. The proportion of SDHB CNAs that were whole gene deletions (5.6%), exon 1 deletions (49%) and exon 3 deletions (14%) was similar to that in other published cohorts,8 28 29 except for cohorts from the Netherlands where exon 3 deletions predominate.30 31 We found 83% (5/6) of SDHC CNAs were exon 6 deletions, and all three SDHD CNAs in our series were exon 4 deletions.
Intragenic mutations
Three hundred and forty-four probands and 436 of their relatives harboured an intragenic mutation in SDHB/SDHC/SDHD (table 1). Forty-five intragenic mutations in 134 probands were reported previously.10 The ratio of mutation classes among probands was similar to that reported previously10 (44% missense, 15% nonsense, 13% splice-site, 15% frameshift, 0.5% inframe deletions and 12% large CNAs). There were a number of recurrent mutations, for example, SDHB splice-site c.72+1G>T and SDHD missense c.242C>T (p.Pro81Leu) mutations accounted for 20% of probands, and the 10 most common mutations accounted for 48% (see online supplementary figure 3).
List of all intragenic mutations found in SDHB, SDHC and SDHD
Clinical presentation
Of 297 probands with a SDHB/SDHC/SDHD mutation and detailed clinical information, the presenting features were predominantly PPGL and HNPGL, though the frequency varied by gene (see online supplementary table 3). Three hundred and seventy-four of the 454 non-proband SDH mutation carriers with detailed clinical information were asymptomatic at testing.
Age-related tumour risks
Tumour risks were estimated by Kaplan-Meier analysis for all SDHB/SDHD/SDHC mutation carriers, probands only and non-probands only for SDHD non-proband carriers only those with paternally inherited mutations). For SDHB, retrospective cohort analysis was also performed using all samples combined (probands and non-probands), probands only and using non-probands only.
Kaplan-Meier analysis (and log-rank testing) of probands and non-probands (figure 1) revealed higher penetrance for symptomatic tumours in SDHD than in SDHB mutation carriers (P=0.032), increased age-related risk of HNPGL in SDHD than SDHB mutation carriers (P<0.0001) and higher risk of PPGL in SDHB than SDHD (P=0.00019). SDHB carriers were more likely to develop malignant disease (P=0.0043). These findings remained significant when the previously reported 358 patients10 were excluded from the analysis (data not shown).

Penetrance of for clinically diagnosed disease in proband and non-proband SDHB, SDHC and SDHD mutation carriers with 95% CI shaded. HNPGL, head and neck paraganglioma; PPGL, phaeochromocytoma and paraganglioma.
In 34 SDHC mutation carriers with detailed clinical information, 19 were clinically affected (15 HNPGL, 3 PPGL and 1 had both HNPGL and PPGL). One patient with HNPGL had local spread and malignant features. Age-related risks of symptomatic PPGL/HNPGL in SDHC were similar to that of SDHD. Compared with SDHB mutation carriers, SDHC carriers had a lower risk of PPGL (P=0.02 and P=0.06 before and after Bonferroni correction) and a higher risk of HNPGL (P<0.001 after Bonferroni correction for three comparisons, log-rank test) (figure 1).
Overall tumour risks will be inflated by including both probands and non-probands (as probands always have the disease) so we re-estimated the cumulative risks in non-probands only. Using Kaplan-Meier analysis, the estimated risk of PPGL/HNPGL at age 60 years in SDHB, SDHD and SDHC mutation positive non-probands was 21.8% (95% CI 15.2% to 27.9%), 43.2% (95% CI 25.4% to 56.7%) and 25% (95% CI 0% to 57.0%), respectively. Nine non-probands (all SDHB) developed malignant disease, and the risk of malignant disease at age 60 years in non-proband SDHB mutation carriers was 4.2% (95% CI 1.1% to 7.2%).
As expected, estimates of penetrance from the retrospective cohort analysis of a combined sample of probands and non-probands were lower than the above estimates from Kaplan-Meier analysis as the former method provides some adjustment for ascertainment bias. Under the retrospective cohort analysis, the predicted penetrance of PPGL/HNPGL in SDHB mutation carriers (probands and non-probands) by age 60 years and age 80 years was 23.9% (95% CI 20.9% to 27.4%) and 30.6% (95% CI 26.8% to 34.7%), respectively (see figure 2), similar to the Kaplan-Meier estimates in non-probands.

Penetrance of clinical disease in SDHB mutation carriers by age 60 years, as calculated by different statistical techniques and in different subpopulations. HNPGL, head and neck paraganglioma; PPGL, phaeochromocytoma and paraganglioma; RCC, renal cell carcinoma.
Male SDHB mutation carriers have a higher age-related penetrance of PPGL/HNPGL (P=0.0034) and PPGL (P=0.0079), compared with women (see online supplementary figure 4), as calculated by Kaplan-Meier analysis and log-rank testing.
Other tumours
Previously,10 we reported 12/358 patients had renal tumours (11 SDHB and 1 SDHD), and the updated cohort contains an additional four cases (all SDHB). Of 751 patients with detailed clinical information (584 SDHB, 33 SDHC and 134 SDHD), 15 (2.6%) SDHB carriers and 1 (0.7%) SDHD carrier had a renal tumour. The risk of developing a renal tumour by age 60 years in SDHB mutation carriers was 4.2% (95% CI 0.46% to 7.8%) by Kaplan-Meier analysis of non-probands, 4.71% (95% CI 1.65% to 7.7%) by Kaplan-Meier analysis of all SDHB mutation carriers and 5.6%/3.2% (male/female) by retrospective cohort analysis of all SDHB mutation carriers. No further thyroid tumours were found beyond the three described previously in SDHB mutation carriers,10 giving an estimated penetrance of thyroid tumours by age 60 years of 1.5% (95% CI 0.0% to 3.1%) (calculated by Kaplan-Meier analysis of all SDHB mutation carriers). Other rare tumours included: (A) SDHB carriers: pituitary adenoma, parathyroid adenoma and pulmonary carcinoid tumour; (B) SDHC: pituitary adenoma, gastrointestinal stromal tumour (GIST); and (C) SDHD: a pituitary tumour.
Genotype–phenotype correlations
Structural prediction analysis
SDHB and SDHD missense mutations were analysed using DUET24 to predict their effect on protein stability and mCSM-PPI to predict their effect on succinate complex formation22 (table 2).
DUET score for all SDHB and SDHD missense mutations described in this cohort and for rare missense variants with no or unknown pathogenicity
SDHB/SDHD variants with no or uncertain pathogenicity were predicted by DUET to have little effect on either protein stability and protein complex formation (average ΔΔG −0.15 Kcal/mol) in contrast to disease associated mutations (average ΔΔG −1.41 Kcal/mol) (P<0.001 two-tailed t-test).
Of the 21 disease-associated SDHB missense mutations, 20 are predicted to be destabilising, 16 are predicted to destabilise the complex and 4 (p.Cys98Arg, p.Cys101Tyr, p.Cys113Tyr and p.Cys196Tyr) are in metal coordinating cysteines. The most destabilising SDHB missense mutation, p.Ile127Ser, affected an isoleucine residue buried deep in the protein with a strong network of intramolecular hydrophobic interactions32 (figure 3A). The three SDHB mutations not predicted to alter protein stability were all predicted to affect the coordination of an iron–sulphur cluster, either directly or by affecting neighbourhood residues (figure 3B).
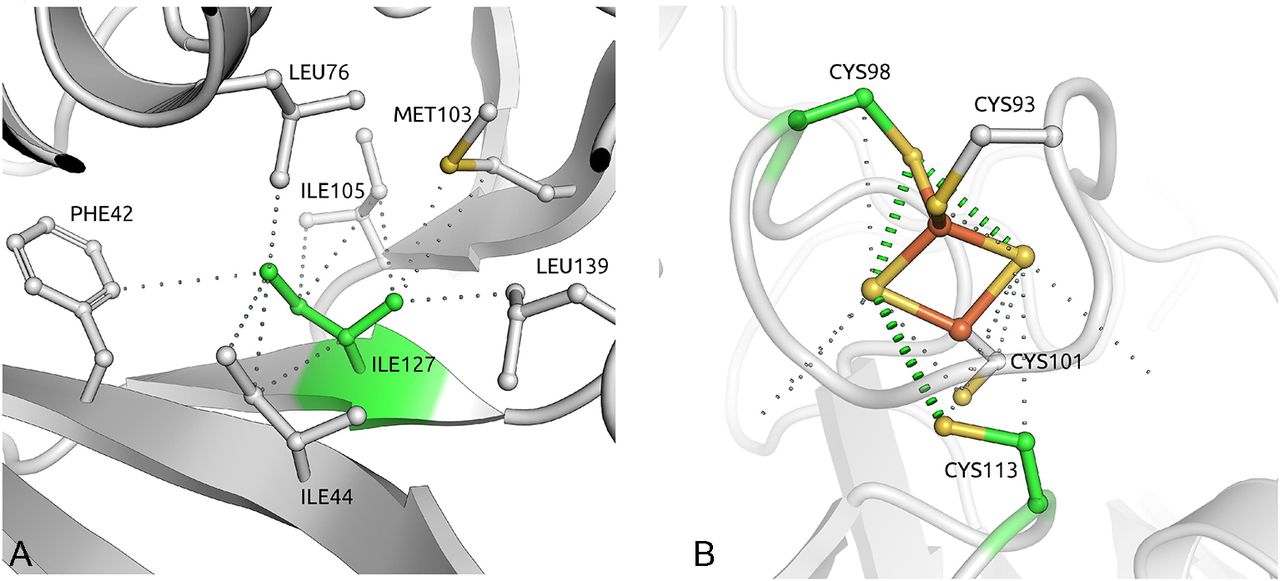
(A) The isoleucine reside at position 127 of SDHB is buried in the middle of the structure, making a network of strong intramolecular hydrophobic interactions. Mutation to serine would introduce a polar residue and disrupt all of these important contacts. (B) One of three iron–sulphur clusters in SDHB, coordinated by four cysteine residues. Four of these (Cys98, Cys101, Cys113 and Cys196) are mutated in our cohort.
Structure–phenotype correlations and mutation specific phenotypes
We confirmed our previous observation10 that the SDHD p.Pro81Leu phenotype is distinct from that of other SDHD mutation carriers, with a low PPGL risk (figure 4). Of 53 individuals with detailed clinical information, 15 were asymptomatic, 37 had HNPGL (two metastatic) and 1 had PPGL (described previously by Yeap et al).16 The p.Pro81Leu mutation is predicted to have a very mild effect on protein stability. Excluding the cases originally analysed by Ricketts et al 10 to create a replication cohort confirmed the lower risk of PPGL in SDHD p.Pro81Leu mutation carriers versus other SDHD mutation carriers (P=0.031, data not shown).

Penetrance of clinical disease in proband and non-proband SDHD p.Pro81Leu mutation carriers versus all other SDHD mutation carriers with 95% CI marked. P values are for the log-rank test comparing the survival distributions of SDHD p.Pro81Leu and all other SDHD mutation carriers. HNPGL, head and neck paraganglioma; PPGL, phaeochromocytoma and paraganglioma.
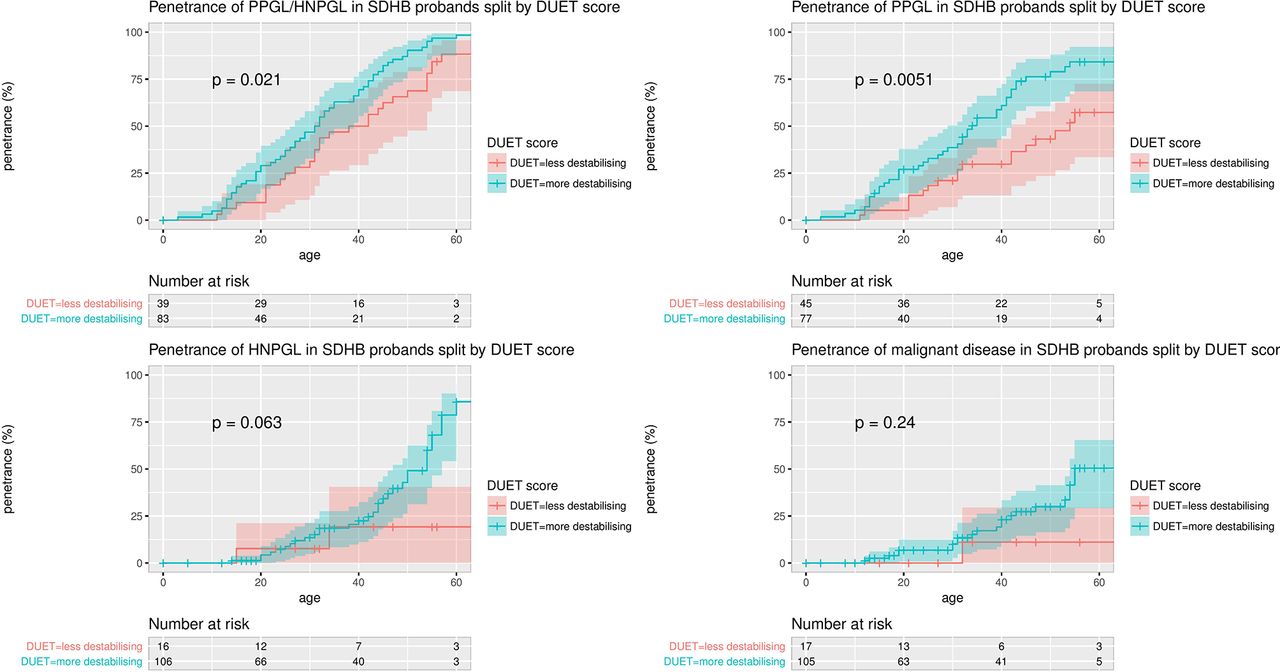
There were no differences in age-related risks of PPGL/HNPGL between missense and truncating variants (data not shown). However, there was a higher overall penetrance for clinical disease (all tumour risk) (P=0.0047) and PPGL risk (P=0.00024) in p.Ile127Ser mutation carriers (the missense mutation with the highest DUET score for predicted protein instability) compared with other missense mutations. Furthermore, using the surv_cutpoint() function in R, which is designed to use the maximally selected rank statistics from the ‘maxstat’ R package to find the optimum cutpoint for continuous variables, the PPGL/HNPGL and PPGL risks were significantly higher for those missense mutations with the most destabilising DUET scores (PPGL/HNPGL penetrance: P=0.0086; PPGL penetrance: P=0.00025). The significance of these differences was preserved when calculated for probands only (PPGL/HNPGL penetrance: P=0.021; PPGL penetrance: P=0.0051, see figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Penetrance of clinical disease in SDHB missense mutation carrier probands, split by with more stabilising (more negative) and less destabilising (less negative) DUET scores. The maximally selected rank statistics from the ‘maxstat’ R package was used to find the optimum cutpoint for DUET score, and the number of probands in the DUET score groups for each analysis is displayed in ‘Number at risk’ tables below each plot. The optimum cutpoints for DUET score were −1.21 for PPGL risk and for PPGL/HNPGL risk, −0.88 for HNPGL risk and −1.02 for malignancy risk. HNPGL, head and neck paraganglioma; PPGL, phaeochromocytoma and paraganglioma.
Discussion
To our knowledge, this is the largest series of SDHB/SDHC/SDHD mutation carriers yet reported (see online supplementary tables 2 and 4) and the previous largest cohort from Buffet et al 28 (363 probands, 269 with SDHB/C/D mutations), which did not report genotype–phenotype correlations. Our large-scale comprehensive genotype–phenotype assessments uncovered a series of novel observations:
Gene-specific and mutation-specific differences in penetrance and expression
We unequivocally confirmed that SDHD mutation carriers had a higher overall penetrance for symptomatic tumours and a higher risk of HNPGL compared with SDHB, whereas SDHB mutation carriers had a higher risk of malignancy and were significantly more likely to develop PPGL. We have confirmed the findings of Jochmanova et al 33 that male SDHB mutation carriers are at higher risk of disease than females (calculated by Kaplan-Meier analysis and log-rank testing). The reason for this observation is not clear. Although it is possible that because asymptomatic female at risk relative might be more likely to come forward for predictive genetic testing than males we note that in our cohort, there were more male than female probands with SDHB mutations (153 male, 110 female), and there were equal numbers of male and female non-probands with SDHB mutations (198 male, 201 female). Of the unaffected at risk relatives without a mutation (relatives of SDHB/D/C mutation carriers), 130 were female and just 103 were male.
The phenotype associated with SDHC mutations has not been well defined. Early reports described benign HNPGLs,5 34 35 but more recent studies have also identified extra-adrenal paragangliomas and more invasive tumours. Bickmann et al 36 and Bourdeau et al 37 both describe SDHC p.Arg133X mutation carriers (a mutation that is also common in our cohort) presenting with extra-adrenal paragangliomas and HNPGLs, both benign and malignant. Our findings of SDHC mutation carriers with extra-adrenal paragangliomas, pheochromocytoma and a case of HNPGL with malignant features are consistent with similar tumour risks with SDHC and paternally inherited SDHD mutations (Lefebvre and Foulkes38 recommend similar tumour screening for SDHC and SDHD mutation carriers).
We replicated our previous finding that SDHD p.Pro81Leu mutation carriers manifest almost exclusively with HNPGL, while other SDHD mutation types predispose to both HNPGLs and PPGLs. From a clinical perspective, it can be proposed that SDHD p.Pro81Leu mutation carriers do not need intensive imaging for PPGL. Although the risk of PPGL in SDHD p.Pro81Leu mutation carriers was not zero, the single patient with a phaeochromocytoma was highly unusual16 and was the only example of tumour development after maternal inheritance of a SDHD mutation. Interestingly, all of the rare cases of tumours in individuals with maternally inherited SDHD mutations have been phaeochromocytomas and none have had a HNPGL.16 39 40 SDHD has a key role in anchoring the SDH complex to the inner mitochondrial membrane, and a truncating SDHD mutation would inactivate the function of the entire SDH complex and be predicted to lead to disordered signalling in the hypoxic gene response pathway and to epigenetic abnormalities resulting from inhibition of enzymes such as prolyl-hydroxylases and ten-eleven translocation enzymes.41–44 The SDHD p.Pro81Leu mutation is predicted not to cause protein instability but to interfere with ubiquinone metabolism/electron transport by changing the folding of helix 1S and by destroying a ubiquinone binding site.10 45 The small risk of PPGL with the p.Pro81Leu mutations suggests that PPGL and HNPGL result from impairment of different aspects of the function of the SDH complex.
Our in silico analysis of the structural effects of SDHB missense mutations revealed a novel genotype–phenotype association in which the missense mutations predicted to have the greatest protein destabilising effects were associated with a higher penetrance and risk of PPGL. If these findings are confirmed, they could be used to stratify tumour surveillance programmes according to individual mutation risks. They could complement other methods, such as tumours studies and in vitro functional studies in human tissues or model organisms such as yeast,46 to elucidate the molecular mechanisms and clinical significance of the mutation.
Other tumours
We also found a variety of rarer tumour types in individuals with SDHB/SDHC/SDHD mutations, including renal cell carcinoma (RCC), GIST, thyroid and pituitary tumours.7 10 47–51 The highest risk for RCC is in SDHB mutation carriers (though they can occur in SDHD and SDHC mutation carriers), and we estimated the risk of RCC by age 60 years in SDHB carriers is 4.7% (95% CI 1.6% to 7.7%) by Kaplan-Meier analysis of probands and non-probands and 5.63%/3.18% (male/female risk) by retrospective cohort analysis.
Although the risk of renal tumours is smaller than for PPGL, if abdominal imaging is being performed for PPGL, it is straightforward to incorporate renal imaging. Our results suggest that thyroid imaging is not indicated if asymptomatic.
Penetrance in non-probands
Accurate knowledge of the natural history of a disease is required for optimum management. For rare diseases, there may be limited numbers of affected individuals, and ascertainment bias in research studies can result in over-representation of extreme phenotypes with overestimation of disease risks. Interestingly, with wider application of SDHB/C/D genetic testing, penetrance estimates for SDHB mutation carriers have declined. Original methods used Kaplan-Meier analysis of both probands and non-probands and produced penetrance estimates such as 75% by age 50 years,7~55% by age 50 years12 and 50% by age 50 years.10 Subsequently, analyses have been published that have controlled for ascertainment bias by excluding probands, producing estimates more like 20% by age 50 years.14 33 An alternative approach has been to study a single large family so that the majority of patients analysed are non-probands, producing penetrance estimates of 35% by age 40 years52 and 26% by age 48.53 More recently, the use of maximum likelihood methods to control for ascertainment bias has resulted in estimates of penetrance as low as 13%,13 9%54 and 21%55 at age 50 years.
The above cohorts have ranged in size from 15 to 344 SDHB mutation carriers. To better understand the prognosis for in individuals undergoing predictive testing, we tested a large (n=584 with detailed clinical information) cohort of SDHB mutation carriers, adjusting for ascertainment bias using two approaches. We estimated overall clinical penetrance and tumour-specific risks by Kaplan-Meier analysis after excluding probands and compared the estimates with those obtained by retrospective cohort analysis in SDHB mutation carriers. We estimated clinical disease penetrance in non-proband SDHB mutation carriers at 50 years, 60 years and 80 years to be 16%, 22% and 44%, respectively, and these were similar with the retrospective cohort analysis estimates: cumulative risk of 24% for PPLG/HNPGL by age 60 years and 31% by age 80 years. One limitation to our analysis was that the unavailability of pedigree structures that would have allowed us to look for potential parent-of-origin effects and to fully adjust for ascertainment.
The recent demonstrations of lower tumour risks raise important questions regarding follow-up for asymptomatic SDHB mutation carriers. Blood/urine biochemical analysis to detect metanephrines is inexpensive, but the risk of non-secretory and malignant PPGL/HNPGL and renal and GIST tumours would argue for regular imaging. The Endocrine Society guidelines56 recommend annual biochemistry with urine or plasma metanephrines and 2 yearly cross-sectional imaging using either CT/MRI of skull base, neck, thorax abdomen and pelvis. It should be noted that the our tumour risk estimates are for clinically apparent disease and are censored at the time of genetic testing. Subsequently, asymptomatic mutation carriers will have undergone biochemical and radiological surveillance and, though this information was not available, some may have been found to have an asymptomatic tumour. Thus, in a study of 30 SDHx mutation carriers,57 a tumour was detected in one patient after 1 year follow-up from normal baseline imaging. Tufton et al 58 describe a cohort of 65 asymptomatic SDHB mutation carriers undergoing surveillance by MRI. They found that 25% of these patients had a likely SDH-related tumour identified after baseline imaging or after a further 2–6 years of screening. The penetrance estimates for clinically apparent tumours in our study will be lower than those estimated in studies where patients have completed comprehensive screening and follow-up but are comparable with those from other large cohort studies and are consistent with more recent, although smaller, studies (see above) that have concluded that SDHB mutations are not associated with a high lifetime risk for clinically apparent tumours. Although the non-inclusion of asymptomatic screen-detected lesions might be viewed as a limitation of the study design, it does avoid the uncertainties regarding whether asymptomatic tumours would become symptomatic, and the risk estimates for clinically apparent tumours are highly relevant to patients wishing to know their lifetime risks of having a symptomatic tumour.
In the light of the robust data from this and other studies demonstrating lower than previously estimated tumour risks in SDHB non-proband mutation carriers, there is a pressing need to continue to systematically collect data on the outcome of surveillance programmes. It is also highly relevant to develop strategies to enable patient stratification (eg, individuals at high risk because of specific mutations or family history/genetic modifiers) and to enable non-invasive early detection of tumours.
Acknowledgments
We would like to thank the patients and the many clinicians and clinical laboratory scientists who contributed to this research. We apologise to researchers whose work we have not cited because of space limitations.
References
Footnotes
Contributors ERM conceived, designed and supervised the study. KAA collected the data and performed the Kaplan-Meier analysist. DBA and DEVP produced the in silico structural prediction analysis. DRB and ACA performed the retrospective cohort analysis calculations. The remaining authors clinically characterised the patients at their respective centres. All authors were involved in drafting the article or revising it critically for important intellectual content and in the final approval of the version to be submitted. KAA and ERM wrote the first draft of the paper and are responsible for the overall content as guarantors.
Funding This work was supported by the East Anglian Foundation Programme (to KAA), the University of Cambridge (KAA), NIHR Cambridge Biomedical Research Centre (KAA and ERM), Cancer Research UK Cambridge Cancer Centre (ERM), European Research Council Advanced Researcher Award (ERM), British Heart Foundation (EM), NIHR Senior Investigator Award (ERM), Newton Fund RCUK-CONFAP Grant awarded by The Medical Research Council (MRC), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) (to DEVP and DBA), NHMRC CJ Martin Fellowship (APP1072476) and Jack Brockhoff Foundation (JBF 4186, 2016) (to DBA), Instituto René Rachou (CPqRR/FIOCRUZ Minas) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (to DEVP), Health Research Board Ireland (RTC) and Cancer Research – UK grant C12292/A20861 (to ACA). The University of Cambridge has received salary support in respect of EM from the NHS in the East of England through the Clinical Academic Reserve. The views expressed are those of the authors and not necessarily those of the NHS or Department of Health.
Disclaimer The funding agencies had no role in study design, data collection or analysis or writing of the article.
Competing interests None declared.
Patient consent Obtained.
Ethics approval South Birmingham Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.