Article Text

Abstract
Non-therapeutic research with imminently dying patients in intensive care presents complex ethical issues. The vulnerabilities of the imminently dying, together with societal disquiet around death and dying, contribute to an intuition that such research is beyond the legitimate scope of scientific inquiry. Yet excluding imminently dying patients from research hinders the advancement of medical science to the detriment of future patients. Building on existing ethical guidelines for research, we propose a framework for the ethical design and conduct of research involving the imminently dying. To enable rapid translation to practice, we frame the approach in the form of eight ethical questions that researchers and research ethics committees ought to answer prior to conducting any research with this patient population. (1) Does the study hypothesis require the inclusion of imminently dying patients? (2) Are non-therapeutic risks and burdens minimised consistent with sound scientific design? (3) Are the risks of these procedures no more than minimal risk? (4) Are these non-therapeutic risks justified insofar as they are reasonable in relation to the anticipated benefits of the study? (5) Will valid informed consent be obtained from an authorised surrogate decision maker? (6) How will incidental findings be handled? (7) What additional steps are in place to protect families and significant others of research participants? (8) What additional steps are in place to protect clinical staff and researchers? Several ethical challenges hinder research with imminently dying patients. Nonetheless, provided adequate protections are in place, non-therapeutic research with imminently dying patients is ethically justifiable. Applying our framework to an ongoing study, we demonstrate how our question-driven approach is well suited to guiding investigators and research ethics committees.
- ethics- research
- death
- risk assessment
- tissue and organ procurement
Data availability statement
Data sharing not applicable as no data sets generated and/or analysed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Non-therapeutic research with imminently dying patients in the intensive care unit (ICU) presents complex ethical issues. Systematic research of this kind is hindered by several ethical challenges, including difficulties obtaining consent for research participation, the ICU as a research environment, the potential for study interventions to interfere with routine end-of-life care and the vulnerability of imminently dying patients.1–4
In this article, we describe a systematic approach to managing ethical issues in research with imminently dying research participants in the ICU. To enable rapid translation to practice, we provide a checklist of eight ethical questions that researchers and research ethics committees must satisfactorily answer prior to conducting any research with this patient population. To illustrate the advantages of this question-driven approach, we apply it to our Canadian research programme, Neurologic Physiology after Removal of Therapy (NeuPaRT). We demonstrate that, provided adequate protections are in place, research with imminently dying research participants is ethically justifiable.
The NeuPaRT study
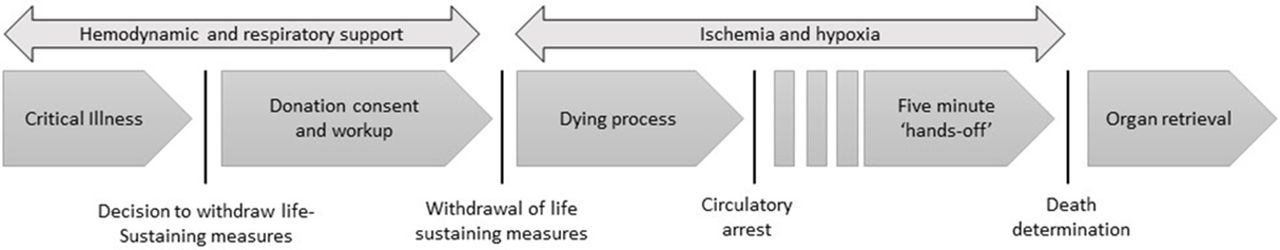
In controlled organ donation after circulatory determination of death (cDCDD), patients with poor prognosis undergo withdrawal of life-sustaining measures (WLSM) and progress to circulatory arrest. Following initiation of WLSM and continuing postmortem, organs suffer ischaemic damage and may become unsuitable for transplantation; hence, surgical organ retrieval begins as soon as possible following the donor’s death. To preclude the possibility of cardiac autoresuscitation, death is declared after a mandatory 5 min ‘hands-off’ period following circulatory arrest, after which organ retrieval commences5 (figure 1).
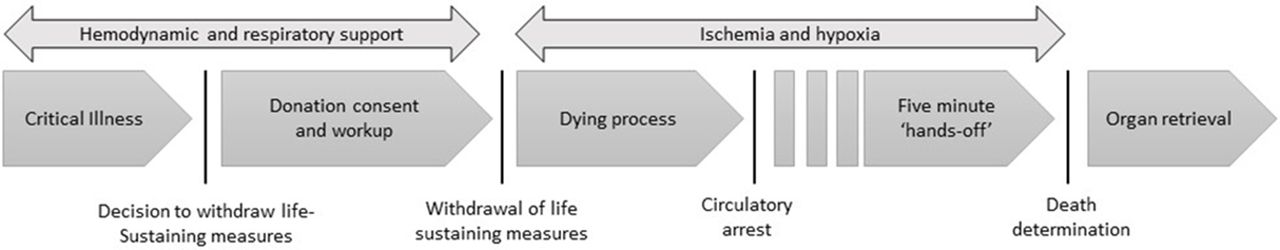
The process of controlled organ donation after circulatory determination of death begins with a decision to withdraw life-sustaining measures and ends with postmortem organ recovery.
Current cDCDD protocols assume permanent loss of brain activity within the ‘hands-off’ period.6 While this assumption is rooted in physiological principles, lack of confirmatory data from patients contributes to unease among a proportion of stakeholders.7 8 Concern stems from the remote possibility that some donors could retain residual brain function during organ retrieval, exposing them to harm and violating the dead donor rule—the injunction that organ retrieval cannot be the cause of donor death.9
By documenting when brain activity ceases relative to circulatory arrest after WLSM in the ICU, NeuPaRT will provide data to inform the timing of death determination in cDCDD (figure 2). Specifically, we use transcranial Doppler (TCD) to measure cerebral blood flow velocity, 10-20 international system video electroencephalogram (v-EEG) to capture cerebral electrical activity, somatosensory evoked potentials (SSEPs) to measure cortical activity, brainstem auditory evoked potentials (BAEPs) to measure brainstem activity, and pre-existing routine monitoring to record arterial pulse pressure, cardiac electrical activity and oxygen saturation (online supplemental material). While these interventions will provide valuable scientific information, their use in research with imminently dying patients poses ethical challenges (table 1).
Supplemental material

{kind=link}
{kind=link}
Neurologic Physiology after Removal of Therapy involves 30 min of baseline recording prior to withdrawal of life-sustaining measures and 30 min of recording following asystole. BAEP, brainstem auditory evoked potential; SSEP, somatosensory evoked potential; TCD, transcranial Doppler; v-EEG, video electroencephalogram; WLSM, withdrawal of life -sustaining measures.
Neurologic Physiology after Removal of Therapy: study details
Materials and methods
Using existing ethical guidelines for research with human participants, we describe a systematic framework for the ethical design and conduct of research involving imminently dying patient participants in the ICU. To enable rapid translation to practice, we frame the approach in the form of eight ethical questions that researchers and research ethics committees ought to answer prior to conducting any research with imminently dying patients. The scope of this paper is limited to ethical issues and does not include an analysis of legal issues. Researchers should be aware of and follow legal requirements for surrogate decision-making and research.
Research with the dying patient
Most studies involving dying patients take place in the context of palliative care, which encompasses patients who have weeks or months left to live. Ethically conducted research in palliative care includes both therapeutic interventions which offer the prospect of direct benefit, as well as non-therapeutic research involving observation, interviews or other non-therapeutic interventions.10 11 Patients in palliative care can often give informed consent and may also benefit directly from research participation.12
Research on imminently dying patients—that is, those who have hours or minutes left to live—differs in important respects from research in palliative care. Researchers are typically unable to obtain first-person consent, nor are patients likely to benefit from participation. While studies involving imminently dying patients are not unknown,13 research intruding into the dying process presents challenges unfamiliar to most researchers. Together, these features distinguish research with the imminently dying from other areas of inquiry.
Patients who are imminently dying are vulnerable because they are at an identifiably increased risk of greater or additional wrongs in research.14 Specifically, they are at risk of autonomy wrongs, such as being treated as mere means to research ends. They are liable to justice-related wrongs through exploitation, or from institutional or professional gatekeeping unfairly preventing research participation. Finally, they are prone to welfare wrongs if the research impedes the provision of end-of-life care.
Suggesting that the vulnerability of the imminently dying precludes research participation is paternalistic and—as with palliative patients—may itself represent an unjust exclusion from research. Furthermore, excluding this population from research would compromise future patients (and organ donors) who stand to benefit from greater understanding of the dying process. While the vulnerability of the imminently dying complicates the application of accepted ethical principles guiding the design and conduct of research, there is no compelling reason why they should be excluded from research provided adequate protections are in place.
All research involving human participants is guided by the ethical principles of justice, respect for persons and beneficence.15 16 These principles entail normative guidelines for the protection of research participants (table 2).
Principles of research ethics and entailed normative guidelines
Further to the usual protections afforded research participants, additional protections for incompetent patients in research are required.15–17 These additional protections may include:
Answering the study question must require the inclusion of these vulnerable participants; that is, their inclusion in research must not be solely for administrative convenience.15–17
The risks of non-therapeutic study procedures must be no more than ‘minimal risk’.16–18
Prospective consent for research participation must be obtained from an authorised surrogate decision maker familiar with the patient’s prior expressed wishes (if any), values and interests.15–17
Finally, recent scholarship in research ethics highlights the need to ensure protections for ‘bystanders’, that is, people who are not research participants but who are nonetheless impacted by research.19
The above normative precepts suggest eight ethical questions that must be addressed prior to conducting any non-therapeutic research with the imminently dying (table 3). To inform investigators and research ethics committees considering research with this population, we describe our answers to these questions in the context of the NeuPaRT study.
Ethical checklist for research with imminently dying patients in the intensive care unit
Results and discussion
Does the study hypothesis require the inclusion of imminently dying patients?
Justice requires that the burdens and benefits of research participation are distributed equitably. Vulnerable participants should not be included in research merely as a matter of convenience. Hence, justifying the inclusion of vulnerable people demands a compelling reason why a study question can only be answered with their inclusion.
To inform the practice of cDCDD, NeuPaRT seeks data on the temporal relationship between cessation of circulatory and neurological activities during the dying process. This question can only be addressed in a controlled ICU environment with patients who are representative of the relevant donor population: imminently dying patients undergoing WLSM.
Are non-therapeutic risks and burdens minimised consistent with sound scientific design?
The ethical principle of beneficence requires, inter alia, that the risks and burdens from non-therapeutic procedures are minimised consistent with sound scientific design. To minimise intrusiveness while balancing the need to maximise the contribution of each patient, any given NeuPaRT participant undergoes no more than two of the four non-therapeutic study procedures. Further, we make use of clinical monitoring already in place where possible, thereby reducing research-related risks and burdens on participants.20 Limiting the number of non-therapeutic procedures that any one patient may undergo and, where possible, using clinical monitoring already in place, minimises risks and burdens to participants while allowing sufficient data to be collected to answer the study question.
The set-up of research equipment and any impact on the medical care of the patient are important potential risks and burdens that must be minimised consistent with sound scientific design. Depending on researcher availability, site location and data collection procedures, non-therapeutic equipment set-up takes up to 1 hour. Contingent on the scheduled time of WLSM, this sometimes calls for a delay before initiation of WLSM. It is conceivable that delayed withdrawal could prolong suffering in patients who retain any degree of consciousness. Furthermore, it is crucial that the presence of researchers does not interfere with routine care or otherwise disrupt interactions between patient, family and significant others and staff.
To minimise risks to welfare and mitigate any changes to the dying process for NeuPaRT participants, the standard of care for patients undergoing WLSM is followed. Routine preparation for withdrawal continues during equipment set-up, including administration of analgesic and anxiolytic medications. There are no restrictions on clinical staff activities. Researchers do not participate in care and are not present in the room after study-related equipment set-up. The impact of study participation on end-of-life care is comparable to other accepted procedures undertaken for the benefit of others, such as preparation for organ donation.
Are the risks of non-therapeutic procedures no more than minimal risk?
Unlike study interventions that may directly benefit participants, procedures administered without therapeutic warrant are subject to a threshold of permissible risk.20 Beneficence requires that any non-therapeutic risks faced by vulnerable participants are minimal. In Canada, ‘minimal risk’ is defined as the risks encountered in the daily lives of the study population.17 The definition of minimal risk varies among jurisdictions and researchers should consult local ethical requirements. Ensuring non-therapeutic risks are minimal is an ethical imperative because competent patients can decide for themselves the research-related risk they are willing to undertake, while incompetent patients cannot. Further, researchers must ensure there are adequate procedures in place for the protection and storage of data and biological samples.
Non-therapeutic components of the NeuPaRT protocol include the addition of neurological monitoring and, in a subgroup of participants, auditory or electrical stimuli at intervals throughout the dying process. The study involves four non-therapeutic procedures, of which no participant undergoes more than two: continuous v-EEG, TCD, SSEPs and BAEPs. To reduce interference, all neurological monitors are positioned out of the way of family and staff and set to ‘comfort’ mode so that sound and display are off. Where this is not possible (BAEPs and SSEPs), monitors remain out of view of family (table 4).
Monitors employed in Neurologic Physiology after Removal of Therapy
Video electroencephalogram
Full-montage v-EEG enables determination of the time at which cerebral electrical activity ceases. Commonly used with critically ill patients in intensive care,21 continuous v-EEG involves a standardised placement of EEG electrodes using the 10-20 international system to allow for adequate coverage of the head.22 [1] V-EEG is non-invasive, passively measuring cortical brain activity. While there are no risks associated with this procedure, the video component may be perceived as intrusive.
EEG’s video component is necessary because EEG is sensitive to artefact. Interpretation of EEG signals requires video recordings to eliminate confounders, such as staff or family and significant others touching the patient or their bed (which they are free to do). The sensitivity of dying process may provoke uneasiness at the prospect of recording. Available recommendations24–26 on the ethical use of videography in research were of limited use in this case, where the purpose and scope of recording is highly circumscribed.
To allay privacy concerns and limit intrusiveness, the EEG camera captures only the patient’s bed. Sound is not recorded. Recordings are deidentified and stored in a secure database. If data contamination is suspected, only the relevant frames of video are reviewed. Only two researchers (TG and DD) have access to the recordings, which will be destroyed 15 years after study completion as per local institutional requirements.
Transcranial Doppler
TCD is a non-invasive procedure which uses ultrasound probes to measure blood flow velocity in intracranial vessels. Once signals are identified by study personnel, two probes are affixed over the temples using a head harness. TCD allows for determination of the time of cessation of cerebral blood flow relative to circulatory arrest.
While there are no physical risks associated with this procedure, the addition of the head harness may be uncomfortable for participants who retain a degree of consciousness. Although this is unlikely in sedated patients, consistent with standard of care a bedside nurse monitors the patient for discomfort and administers analgesics as needed.
BAEPs and SSEPs
BAEPs and SSEPs are used in our study to determine the cessation of brainstem and cortical function, respectively, and provide data to interpret whether cerebral electrical activity measured via v-EEG represents brain function as opposed to activity. BAEPs use a series of clicks at a volume of 60 dB delivered through an earpiece,27 and SSEPs use an electrical stimulus administered to the median nerve in the wrist.28 While no participant undergoes both SSEPs and BAEPs, each could cause discomfort in residually aware participants.
Phenomenologically, conscious adults experience BAEPs at a volume of 60 dB as similar to a conversation between two adults sitting 1 m apart. SSEPs are like dull but persisting electrostatic shocks and described as mildly uncomfortable. Twenty-four healthy volunteers studied locally rated the overall mean of pain of SSEPs as 2.51/10 (SD=2.04), with 1=no pain, 5=moderate pain, 10=severe pain (Loretta Norton, personal communication, 28 April 2022). We limit the obtrusiveness of stimuli by presenting to only one ear or one wrist and follow the American Clinical Neurophysiology Society guidelines.28
While v-EEG and TCD pose no risks to participants, BAEPs and SSEPs arguably pose welfare risks in the form of discomfort or anxiety. Since participants in our study are sedated, this is unlikely; however, the possibility cannot be eliminated. Were evoked potentials to cause a patient distress (as indicated by the patient’s behaviour, heart rate or blood pressure), we would immediately discontinue the procedure.
Are these non-therapeutic risks justified insofar as they are reasonable in relation to the anticipated benefits of the study?
Beneficence demands that study risks are reasonable in relation to anticipated benefits. While the impact of NeuPaRT participation on end-of-life care is minimised and the risks posed do not obviously exceed the minimal risk threshold, still they must be justified with respect to the social value of the study.
The NeuPaRT study addresses scientific questions central to organ donation. By failing to confirm permanent loss of brain activity, the current approach to cDCDD may fail to protect donors from harm. Conversely, should cessation of brain activity precede circulatory arrest, the current approach may deny donors the opportunity to bequeath optimally viable organs. The comprehensive data collection procedures we employ are necessary to rigorously demonstrate the temporal relationship between cessation of brain and circulatory activity in cDCDD candidates. This may dispel lingering doubts about residual brain function and the appropriate timing of death determination in cDCDD. The benefits to be gained from our study—better outcomes for organ recipients, potentially increased quality and quantity of transplantable organs, protection of future donors—are substantial. As the risks to participants are minimal and the social value of the study is high, we conclude it has an acceptable benefit-harm profile.
Will valid informed consent be obtained from an authorised surrogate decision maker?
When the prospective research participant is incompetent, respect for persons requires consent from an authorised surrogate decision maker familiar with the patient’s prior expressed wishes, values and interests. Yet the ICU environment and the difficult circumstances attending a patient’s illness or injury are obstacles to informed consent.3 4 Potential impediments include therapeutic misconception (attributing clinical intent to research activities), undue influence on the part of treating physicians and the degree to which surrogates can make informed decisions in an unfamiliar environment while experiencing distress or confusion about complex health information.4 29–31
Clinical staff are valuable partners for deciding whether to approach family and significant others about patient participation. Before approaching a surrogate decision maker, researchers confer with nursing staff and inquire as to whether they think it appropriate. If nursing staff are uncertain, researchers discuss the possibility with a social worker familiar with the family. To avoid further distressing families, researchers defer to the judgement of staff.
Feelings of reciprocity towards clinical staff could contribute to instances of invalid consent.3 Additionally, surrogates may fear that refusal will negatively impact quality of patient care.30 For these reasons, approaches for consent are initiated by our researchers, and only after a decision has been taken on WLSM and (when applicable) organ donation. In the consent process, surrogates are informed that refusal of study participation will not impact patient care.
Because the addition of neurological monitoring alters the appearance of the patient, transparency is important to ensure the surrogate is informed. To allow surrogates to visualise what participation entails, the study’s letter of information includes pictures of research equipment, as well as an image of a person with neuromonitoring on an ICU bed (see online supplemental file 1).
Therapeutic misconception occurs when the person providing informed consent attributes clinical intent to non-therapeutic research interventions.32 Surrogates may not perceive a distinction between routine clinical care in the ICU and research procedures. Having a researcher who is not part of the clinical team approach the family and significant others for consent combats therapeutic misconception by highlighting the distinction between routine care and research.
Some of the patients in the study go on to become organ donors. In these cases, surrogates could conflate procedures required for organ donation with research interventions. For this reason, researchers allow the donation team to approach surrogates first. Additionally, the consent document makes clear both that data collection will not interfere with donation protocol and that withdrawal from the study in no way affects the prospects of donation.
How will incidental findings be handled?
An incidental finding is a discovery about a research participant made during research that is outside the scope of the study, and which could indicate a change in care.17 Any research in which the discovery of incidental findings is foreseeable requires plans be in place for their management.33 One worry raised by the research ethics board at our site concerned those participants proceeding to cDCDD: how would we manage the discovery of neurological activity after the ‘hands-off’ period following circulatory arrest required before death determination?
For two reasons, this eventuality cannot arise in our study. First, cDCDD follows a well-defined protocol with which our study does not interfere. Second, to provide families and significant others with privacy, researchers do not observe neuromonitors in real time. All data analyses and interpretation take place offline. Hence, no incidental findings will arise.
What additional steps are in place to protect families and significant others of research participants?
NeuPaRT is a useful illustration of when the scope of research ethics guidelines can fail to account for impacts on third parties. Families and significant others of patients involved in our study are research ‘bystanders’: non-participants who may be impacted by research activities.19 Already at risk of psychological harm from traumatic experience in the ICU,34 and sometimes in a state of anticipatory grief,29 family members are themselves vulnerable. While not encompassed by research ethics guidelines, it is imperative to minimise the impact of changes to end-of-life processes which could adversely impact families.
In our study, participants’ families and significant others are encouraged to remain at the bedside as the research is conducted. There are no restrictions on their interactions with the patient or staff. If required, families are supported through the most appropriate means, including social services and spiritual care.
Studies on family experience in organ donation highlight how delayed initiation of WLSM for the purposes of donor workup can be distressing for families.35 36 The lesser delay sometimes required for our study set-up could be experienced similarly. To mitigate impact stemming from delay, families and significant others are informed of the time required for set-up during the consent process. Following clinical determination of death, monitoring continues for an additional 30 min with those participants not proceeding to organ donation. This duration was acceptable to participants’ families in a similar study measuring cardiac activity during the dying process.13
Because several study procedures alter the appearance of participants, families and significant others may find the presence of additional monitors, probes and leads confronting. The images of the study monitoring devices and their placement in the study’s letter of information are thus integral to preparing families. Survey and interview data2 37 and high consent rates from a pilot study using neurological monitoring at end of life (specifically, non-therapeutic EEG) suggest that families do not find neuromonitoring overly invasive.38
Further, family members may perceive study involvement as a benefit for the family. Families may derive meaning from the loss of a loved one by helping to contribute to the production of scientific knowledge.37 Families may find value in their role in fulfilling the prior expressed wish of the patient to participate in research or, more generally, in enabling socially valuable research. To facilitate such meaning-making for families and significant others, we communicate the social value of the research, and we plan to provide families with summary research findings after peer review and publication.
Finally, we believe that researchers should engage families and significant others of patients who have died in the ICU in the study design process and the preparation of consent materials. The direct involvement of families in the research process will help ensure that study procedures are conducted in a way that families and significant others will find acceptable and that their information needs are met.
What additional steps are in place to protect clinical staff and researchers?
An often overlooked aspect of the conduct of research in the ICU is the potential for a study to impact clinical staff and researchers.39 This is a particularly relevant concern in our study, where these stakeholders must navigate an emotionally fraught environment.
ICU clinical staff have extensive experience with end-of-life situations, and healthcare professionals are generally supportive of observational research conducted at the end of life.10 39 Prior to commencing enrolment, we offered a series of presentations on the scope, aims, social value and procedures of the study. Staff received handouts summarising key information, and after each session we allowed time for questions and concerns. A similar study involving imminently dying patients reported approval of the research among staff.2
Researchers involved with studies on the imminently dying could find the experience emotionally draining or otherwise burdensome insofar as they may feel they are inserting themselves into a sensitive and private moment.39 Additionally, logistical challenges (eg, consent with larger families, broaching a difficult topic) are demanding and may cause frustration or unease.
Ensuring that researchers are adequately trained for research in this setting is not only an issue of competency, it protects families and researchers from needless misunderstanding and conflict. We ensure that our researchers are experienced in approaching families and significant others for consent for research in the ICU. They are familiar with both the patient population and the testing techniques involved. Researchers use a consent script that addresses the difficulties families and significant others are facing. This resource is specific to our study and provides researchers with aids for navigating an emotionally sensitive environment.
Conclusion
While research with imminently dying patients in intensive care poses ethical challenges, the NeuPaRT experience demonstrates that such research can be conducted ethically. Our systematic checklist of eight ethical questions to answer before conducting non-therapeutic research with the imminently dying will guide researchers and research ethics committees considering similar research.
Data availability statement
Data sharing not applicable as no data sets generated and/or analysed for this study.
Ethics statements
Patient consent for publication
Ethics approval
The NeuPaRT study was approved by the Health Sciences Research Ethics Board at Western University, London, Canada.
Acknowledgments
The authors thank two anonymous reviewers for their constructive comments and helpful suggestions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @NB_Murphy, @charlesweijer
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY. 17th May 2023.
Contributors CW and NM conceived the project. NM wrote the manuscript with substantive support from CW. All authors agreed to manuscript content and structure prior to drafting. DD, TG, GL, LN, MS and CW reviewed all the drafts and provided substantive comments. The final draft was agreed by all authors. The guarantor of this work, NM, accepts full responsibility for the finished work and controlled the decision to publish.
Funding This study was funded by New Frontiers Research Fund (NFRFE-2019-00759).
Competing interests TG reports grants from the Government of Canada, grants from the Canadian Institutes of Health Research, during the conduct of the research. MS reports grants from Lawson Health Research Institute during the conduct of the research; other from Trillium Gift of Life Network, outside the submitted work. CW receives consulting income from Cardialen, Eli Lilly and Company and Research Triangle Institute (RTI) International.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
↵While we use conventional placements of electrodes, we use fewer. Normal EEG has electrodes 6 cm apart. We use 10 cm placings because measuring between longer distances allows greater ability to see cerebral activity. See Stecker et al.23
Other content recommended for you
- Family experiences with non-therapeutic research on dying patients in the intensive care unit
- Informed consent for functional MRI research on comatose patients following severe brain injury: balancing the social benefits of research against patient autonomy
- Neurologic Physiology after Removal of Therapy (NeuPaRT) study: study protocol of a multicentre, prospective, observational, pilot feasibility study of neurophysiology after withdrawal of life-sustaining measures
- The use of electrophysiological monitoring in the intraoperative management of intracranial aneurysms
- Assessing research risks systematically: the net risks test
- Clinical evoked potentials in neurology: a review of techniques and indications
- Intraoperative evoked potential monitoring for detecting cerebral injury during adult aneurysm clipping surgery: a systematic review and meta-analysis of diagnostic test accuracy
- Intraoperative vascular complications during 2278 cerebral endovascular procedures with multimodality IONM: relationship between signal change, complication, intervention and postoperative outcome
- Are positive experiences of children in non-therapeutic research justifiable research benefits?
- The clinical role of evoked potentials