Article Text

Abstract
Data on all presymptomatic genetic tests for Huntington's disease (HD) in the UK have been collected over the 10 year period since testing became available as a service. A total of 2937 completed tests have been performed up to the end of 1997, 2502 based on specific mutation testing, feasible since late 1993.
A total of 93.1% of these were at 50% prior risk, with a significant excess of females (58.3%); 41.4% of results were abnormal or high risk, including 29.4% in subjects aged 60 or over. The trend in test numbers has currently levelled out at around 500 per year.
Almost all presymptomatic tests are carried out in National Health Service genetics centres, with a defined genetic counselling protocol and with availability now in all regions of the UK. The introduction and establishment of HD presymptomatic testing shows that this form of predictive medicine for Mendelian disorders can be successfully incorporated into National Health Service structures. The comprehensive collection of simple data allows trends in demand and outcomes to be monitored and has also been the foundation for more detailed specific studies. A comparable approach to data collection in other genetic disorders will be important as presymptomatic testing becomes more generally feasible.
- Huntington's disease
- presymptomatic testing
Statistics from Altmetric.com
Huntington's disease (HD), an inherited neurodegenerative disorder of later life causing progressive physical and mental deterioration,1 was the first serious autosomal dominant disorder for which genetic prediction became possible using DNA markers. Following localisation of the gene in 1983 by discovery of a closely linked DNA marker on chromosome 4,2 and the subsequent exclusion of genetic heterogeneity, presymptomatic testing based on the use of linked markers began to be initiated in the UK3 4 and elsewhere,5 6 initially in a research framework and subsequently as a service. Careful protocols for counselling and support were developed,7 8 and close co-operation and standardisation of these internationally has allowed detailed evaluation of the outcomes and psychological effects in the short and medium term.9 10
The identification of a specific mutation for HD in 1993,11 and the recognition that essentially all cases resulted from the same mutational mechanism of trinucleotide repeat expansion, provided a presymptomatic test that was highly accurate12 13 and that could be used without the need for samples from multiple family members that had limited testing based on linked genetic markers. Presymptomatic testing based on mutation analysis is now available as a service in medical genetics centres throughout the UK and in most other developed countries.
The UK Huntington's Disease Prediction Consortium was initiated in 1989, two years after presymptomatic testing had been started in the UK. Its aims were to collect anonymous data on all UK presymptomatic tests for HD, and to provide a forum, through its annual meeting, for discussion of problems and difficult issues arising, both of a general and specific nature. The consortium has primarily been clinically orientated and has not attempted to assess quality or other aspects of laboratory procedures, though it has maintained close contacts with the genetics laboratories involved. All centres involved in presymptomatic testing have participated, giving near complete coverage of data from the outset.
In 1992, the Consortium published preliminary results of its combined experience (up to the end of 1990),14 by which stage 248 completed presymptomatic tests had been undertaken, all based on the use of linked genetic markers, since the discovery of the HD gene and mutation did not occur until 1993. The present paper, containing complete data to the end of 1997, marks 10 years of HD presymptomatic testing in the UK and allows assessment of changes in the field as well as being based on a large body of data.
Developments in gene isolation have made mutation based presymptomatic testing feasible for an increasing number of other dominantly inherited neurodegenerative disorders in addition to HD, including familial prion dementias,15 dominantly inherited Alzheimer's disease,16 and familial motor neurone disease.17 These are mostly very rare and practical experience of presymptomatic testing for them remains limited. Experience with HD presymptomatic testing thus provides a valuable general model for these other disorders, and the systematic process of data collection on all presymptomatic tests for HD provides a particularly complete record of experience of this new form of “predictive medicine”.
Methods
An anonymous computerised database was established to record a standard data set on all completed HD presymptomatic tests. Forms were completed by nominated subjects in each centre and were entered onto the database by the Consortium Coordinator in Cardiff. This basic dataset is summarised in table 1; additional information was recorded over limited periods on specific topics, including prenatal tests and diagnostic tests on symptomatic patients (not included in the basic dataset) and subjects at 25% prior risk (a subset of the total prediction data). No data that would allow identification of subjects were transmitted to the Consortium, all queries being relayed for resolution to the specific centre.
HD Prediction Consortium data set
For direct mutation analysis, the number of CAG repeats in theIT15 gene was estimated as originally described by Gusella et al 11with service modifications subsequently to exclude the adjacent CCG repeat.18 The normal range was classified as up to 30 repeats, with 39 or more repeats as abnormal, the small intervening number being considered as “equivocal”. Since this paper reports all UK results over the period since the HD gene was isolated, there will inevitably have been some minor variations in techniques used.
Results
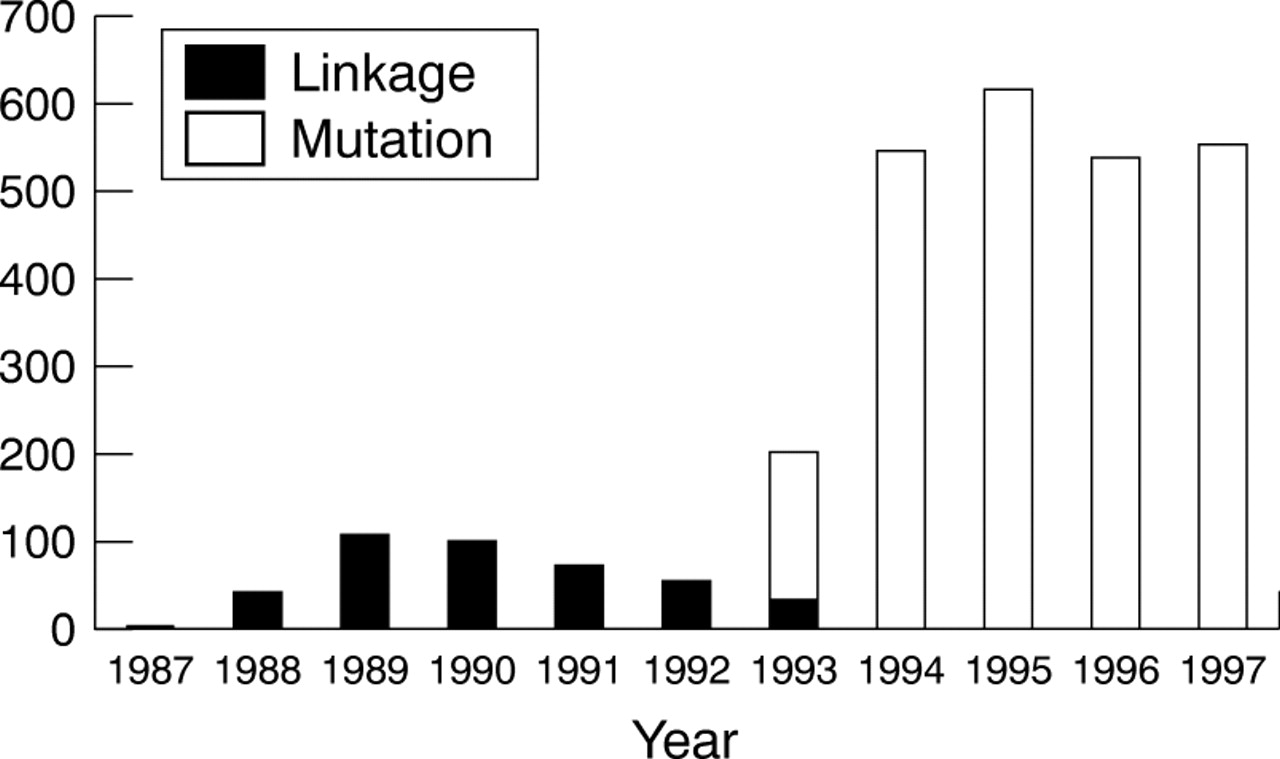
A total of 2937 completed presymptomatic tests for HD had been carried out over the 10 year period to the end of 1997, of which 2502 were based on specific analysis for the HD mutation, 426 were based on linked DNA markers, while nine subjects were tested using both approaches on different occasions. Fig 1 shows the annual trend for these results, divided according to testing by linked markers (up to 1993) and specific mutation analysis (from 1993 onwards).
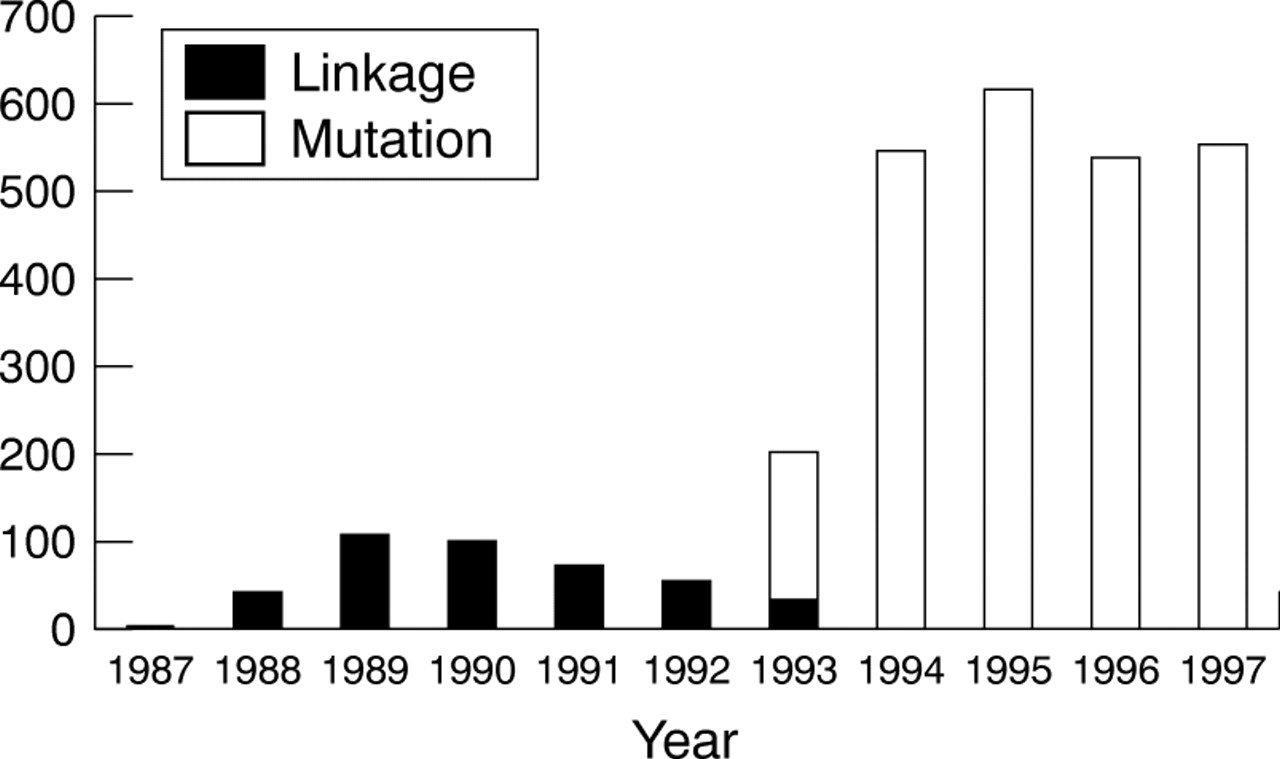
Total number of predictive tests for Huntington's disease in the UK, 1987-1997. Total for year 1998 was 498 (note added in proof).
Table 2 records relevant data on prior genetic risk, outcome, and sex. The age distribution for both normal and abnormal outcomes is shown in fig 2. The mean age for those tested was 36.2 years (median 44.5 years).
UK presymptomatic tests for HD, 1988–1997 (n=2928)

Predictive tests for Huntington's disease divided by age and outcome (mutation testing data only).
Presymptomatic testing for HD is now available throughout the UK. The number of centres had risen to 23 by 1997, while the number of tests carried out by each centre (mean 110) over the five year period 1993-7 is shown in fig 3. It should be noted that a centre was defined as that undertaking the genetic counselling and giving the result rather than the laboratory involved, some laboratories serving several centres, especially in the early stages of the programme. Results were given to those tested by a clinical geneticist in 15 centres, by a clinical geneticist and a genetics nurse specialist jointly in four, by a genetics nurse alone in two, and by a neuropsychiatrist in one centre.19

{kind=link}
{kind=link}
{kind=link}
Numbers of tests carried out by individual genetic centres (1993-1997).
Diagnostic testing results for the HD mutation on symptomatic subjects (only possible following isolation of the gene) were not included in the presymptomatic test data, but were recorded on a summary basis in terms of total numbers for each centre, together with outcomes, and are shown in table 3 for the three years 1995-1997. Variation in the proportion of abnormal results is likely to result from changes in the clinical criteria for requesting molecular analysis.
Molecular diagnostic tests for Huntington's disease in symptomatic subjects, 1995–1997
Discussion
The data presented here reflect the development and experience of presymptomatic testing for HD in the UK, the first serious dominantly inherited disorder for which DNA based presymptomatic testing was feasible. The extent of the data (almost 3000 subjects tested) and its completeness make it a unique record of how an essentially new form of “predictive medicine” has been introduced and established in medical practice, something that is likely to provide valuable lessons for developments in other late onset genetic disorders, where a series of important issues arise which are comparable across different disorders.20 While studies from individual centres have been reported from the UK and elsewhere, which can give detail impossible in a combined dataset such as the present one, the UK Consortium data provide the numbers and the time period required to examine a number of broader issues and trends.
SPECIFIC FINDINGS AND THEIR IMPLICATIONS
The results given in table 2 illustrate several important findings in HD presymptomatic testing. The sex ratio (58% female, p=1.9 × 10-19) confirms previous reports of a female excess among those tested.21 Suggested explanations for this have included a greater involvement with and responsibility of women for areas of life concerned with family and reproductive decisions and a greater willingness of women to face up to difficult decisions and their consequences. The greater uptake of HD testing among females is consistent with data indicating that women are significantly more likely than men to use testing for genes conferring susceptibility to inherited cancer syndromes,22 and it will be of interest to see if a comparable female excess is found in presymptomatic testing for other late onset neurodegenerative disorders.
The age distribution of those tested (fig 2) is of considerable interest. UK and international guidelines advise against the presymptomatic testing of minors for HD and other untreatable adult onset genetic disorders,23 but the number of tests under the age of 20 in this series is small. At the other end of life, the considerable number of tests requested by those aged 50 and over is of note; many such subjects may request testing as much for the benefit of their adult offspring, who may be reaching the point of reproductive decisions, as for themselves. The considerable proportion of abnormal results in these older subjects reflects the extremely wide age at onset of HD and is of practical relevance. In this series, 29% of those aged 60 or over received an abnormal result, in keeping with the increasing recognition that HD may have onset in old age. Such people may perceive themselves no longer to be at serious risk of developing the disorder and may not anticipate the consequences of an abnormal result to the extent of younger subjects, giving extra importance to the process of preparation and genetic counselling before a decision on testing if adverse consequences are to be avoided later. The overall outcome of results in the series, excluding equivocal results, is close to that expected on the basis of age at testing and prior genetic risk when compared with life table studies of age at onset in HD.24 A total of 1018 abnormal results were observed, compared with 989 expected, a small though statistically significant difference (χ2=6.93, p=0.008).
The great majority of those tested (93.1%) had a prior genetic risk of 50% (parent affected). Testing those at 25% prior risk, all of whom had an affected grandparent or other second degree relative, presents particular problems in those cases where the intervening parent is alive and does not wish to be tested or may be unaware that their child has requested it. This group has been the subject of a more detailed study by the Consortium25 over the two year period 1994-1995, and it is relevant that in this study there were only four instances where an abnormal result in the subject requesting testing predicted presence of the HD mutation in a living parent over a period when 85 subjects at 25% prior risk, and 1229 overall, underwent presymptomatic testing.
Up to 1993, when the HD gene and specific mutation were identified, presymptomatic testing was based on the use of closely linked genetic markers, giving an inherent error rate because of recombination and necessitating the sampling of multiple family members. It is not surprising that the need for samples from other family members limited demand during this period, but the rapidity of change to specific mutation testing as a service is notable. When the gene was identified in 1993, most centres contacted those who had already undergone linkage based presymptomatic testing to let them know that a more accurate test for the specific mutation was available, but surprisingly few (nine out of 426) requested repeat testing. The occurrence of a single mutation underlying essentially all cases of HD is highly unusual for genetic disorders; in most conditions the occurrence of numerous different mutations creates technical problems that limit the service use of mutation testing even when the specific gene has been isolated.
It has been suggested that the change from linkage to mutation testing may have been associated with a change in the characteristics of those being tested, but we find no evidence for any difference between those tested by linkage or mutation analysis for sex ratio, prior genetic risk, or outcome (p>0.3 in all cases).
ADDITIONAL STUDIES
The fact that the HD Prediction Consortium database has captured basic information on all UK predictive tests, together with the Consortium's annual discussion meeting, has allowed it to form the starting point for wider studies. The analysis of those tests at 25% prior risk has already been mentioned; a study has also been carried out on prenatal tests for HD26 (data not collected in the core data set). A total of 111 prenatal tests had occurred in the UK during the four years 1994-1997, indicating a low uptake of this form of genetic testing in HD by comparison with presymptomatic testing. Information has also been collected on serious adverse effects (suicide, attempted suicide, or psychiatric disorder necessitating hospital treatment) associated with predictive testing (defined as suicide, attempted suicide, or psychiatric disorder requiring hospital treatment), as part of a world wide study of such events.27 The data confirm that these events are relatively rare in the short term, though long term surveillance will be required to provide an accurate conclusion; it should be noted that all testing in this series was accompanied by full preparation and support by experienced professionals.
FUTURE PROJECTIONS
With data on HD presymptomatic testing thought to be complete for the UK, it is important to examine the trend of demand for testing and attempt to assess what proportion of subjects at risk for HD are actually being tested. Although some studies done before testing became feasible suggested that uptake would be high (up to 80%),28 this was not born out by subsequent experience, which has suggested levels of 5-20%.3 14
The present data are difficult to interpret in this respect, since during the first part of the decade the limitations of linkage based testing excluded many subjects from being tested, while the lack of availability over many areas of the UK resulted in a backlog of those wishing to be tested. We can, however, construct limits for estimates of both the proportion of those at risk that have been tested and what would be expected when a “steady state” is reached.
Estimates of the prevalence of HD in the UK vary from 3-10 × 10-5 ,29 but if we take 7.5 × 10-5 as an approximate prevalence, then the heterozygote frequency has been shown to be around 2.5 × the prevalence30 and the number of subjects at 50% prior risk (half of whom will prove to be unaffected) will be twice this, that is, 37.5 × 10-5.
If this is projected to the UK population of about 40 million (extrapolated from the 1992 England and Wales census data) in the 15-64 years age group, then the number of subjects at 50% risk who might have been eligible for testing is approximately 15 000 (37.5 × 10-5 × 40 × 106). The actual number of tests done on those at 50% risk was 2722, which represents around 18% of those eligible. There are no data which would allow an estimate of the number of at risk subjects in the population who were unaware that predictive testing was a possibility or did not know how to access clinical genetic services, but given the widespread publicity about genetic testing in the media and the increasing activity of lay organisations such as the Huntington's Disease Association it seems unlikely that these represent a large proportion of the total. Since many at risk subjects decline the opportunity of a genetic counselling appointment when it is offered, the overall uptake figure of 18% is broadly comparable with the experience of most of the centres involved.
HEALTH SERVICE IMPLICATIONS
It should be regarded as an example of the adaptability of the UK National Health Service that an entirely new form of medical practice, such as presymptomatic testing for HD, could have been introduced and established on a comprehensive and geographically equitable framework over a relatively short period. The process has been largely taken on board by the existing network of regional medical genetic centres, with their associated laboratories; all parts of the UK now provide the service. We are not aware of any presymptomatic testing in the UK involving the private sector, whether for genetic counselling or laboratory aspects, though it is possible that a very small number of subjects might have sought testing abroad.
It is of relevance that the clinical and genetic counselling aspects of presymptomatic testing should have been undertaken almost exclusively by clinical geneticists and associated staff, rather than by neurologists (the only exception is the Institute of Neurology, London); this is in contrast to the diagnostic testing of symptomatic subjects with suspected HD, where most samples are referred directly to genetics laboratories by neurologists or other specialists. These patterns of practice probably reflect the fact that such clinicians understandably prefer to devote most of their time to the problems of symptomatic patients, while clinical geneticists are better placed to handle the complex and time consuming issues involving those requesting presymptomatic testing, who are generally healthy subjects.
Presymptomatic testing is now becoming feasible for a wide range of late onset, dominantly inherited disorders, and the experience of the UK Consortium and others for presymptomatic testing is proving extremely valuable in those more general applications. Some of the disorders are neurological (for example, prion dementias, mendelian forms of Alzheimer's disease and other dementias, familial motor neurone disease); all are extremely uncommon and presymptomatic testing can be handled using protocols very similar to those in use for HD. An Alzheimer's Prediction Consortium has now been established in the UK, which is particularly looking at issues involving susceptibility testing for the common, non-mendelian forms of the disorder. Familial cancers, including the genetic subsets of breast and colorectal cancer as well as rare tumour syndromes, are emerging as another important group where presymptomatic testing is becoming feasible and increasingly requested.31 While specific studies have been taken on various aspects of presymptomatic testing in familial breast cancer, there is no concerted mechanism, such as reported here for HD, that combines and monitors the experience on a UK basis. We suggest that those involved with presymptomatic testing for these other disorders attempt to collect such data comprehensively and systematically, preferably starting before testing becomes widespread. In this way the important general issues involved in presymptomatic testing for late onset disorders will be able to be examined from a broader evidence base than that provided by a single disorder, such as HD.
Acknowledgments
The individual centres and their contributory staff are listed below*. We thank all the genetics laboratories involved in molecular analysis for HD for the help in the provision and checking of data, Gary Houlihan, Ruth Glew, and Michele Thomas for help in collecting and recording information, Dr Robert Newcombe for statistical advice in relation to age at onset distribution, and the Huntington's Disease association for their encouragement and financial support.
*Aberdeen (Dr Sheila Simpson), Cardiff (Professor Peter Harper), Edinburgh (Dr Mary Porteous), Glasgow (Professor Michael Connor, Dr Brian Smith), Northwick Park, London (Dr Christine Garrett, Ms Lyn Carlton), Institute of Neurology, London (Dr Nicholas Wood, Dr Mary Davis), Manchester (Dr David Craufurd), Newcastle (Professor John Burn), Oxford (Dr Susan Huson, Ms Elizabeth Rosser, Mrs Ruth Glew), Sheffield (Dr Oliver Quarrell), Southampton (Dr Nick Dennis), Nottingham (Professor Sandy Raeburn, Ms Eleanor McGhee), Dundee (Mrs Lorna McLeish), Bristol (Dr Peter Lunt), Liverpool (Ms Caroline Benjamin, Dr Alan Fryer), St George's Hospital, London (Professor Michael Patton, Ms Anne Howick), Northern Ireland (Dr Patrick Morrisson), Cambridge (Dr Richard ffrench-Constant), Guy's Hospital, London (Dr Alison Lashwood), Birmingham (Dr Peter Farndon), Leeds (Dr R Mueller), Exeter (Dr Peter Turnpenny), Leicester (Dr R Trembath).